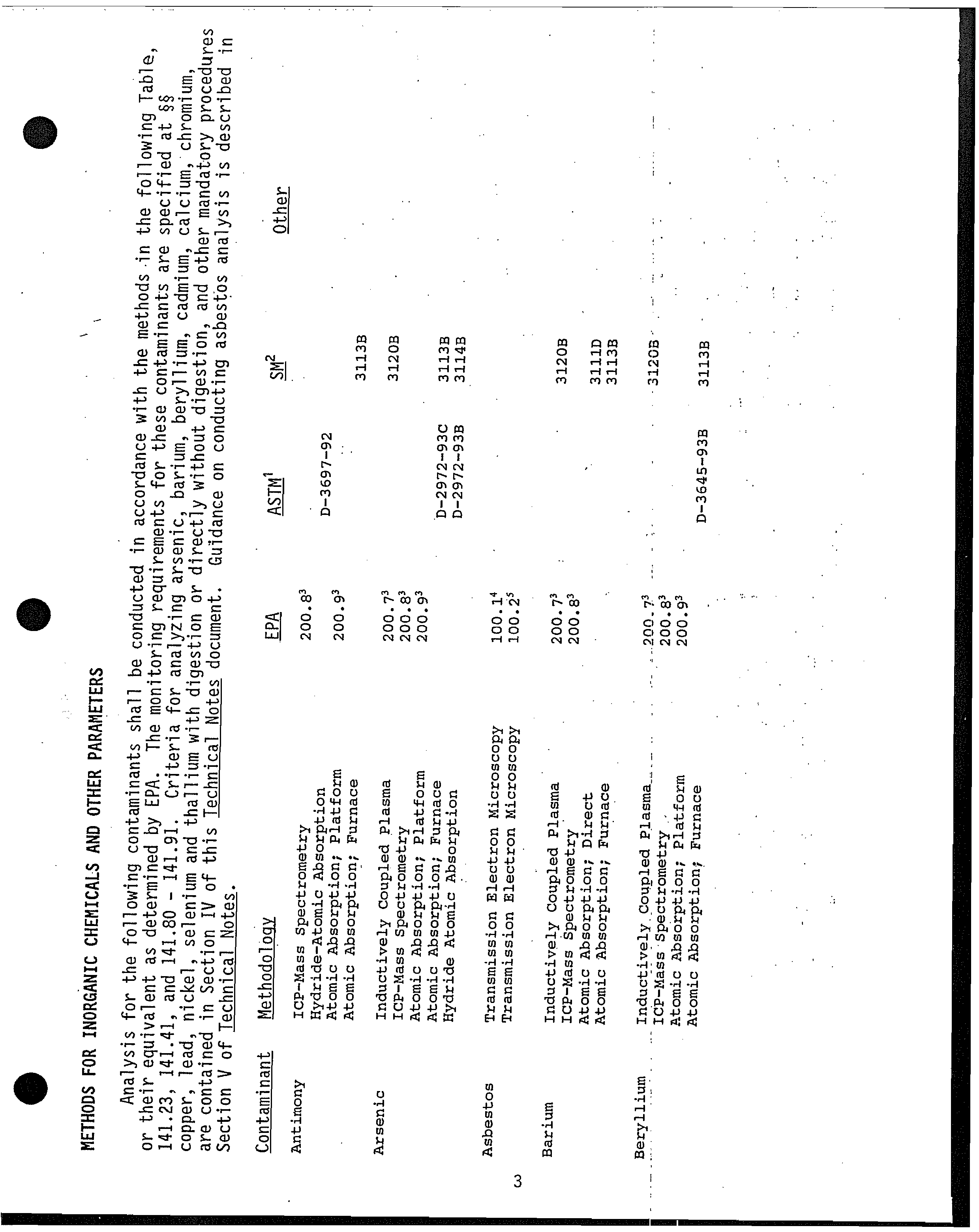
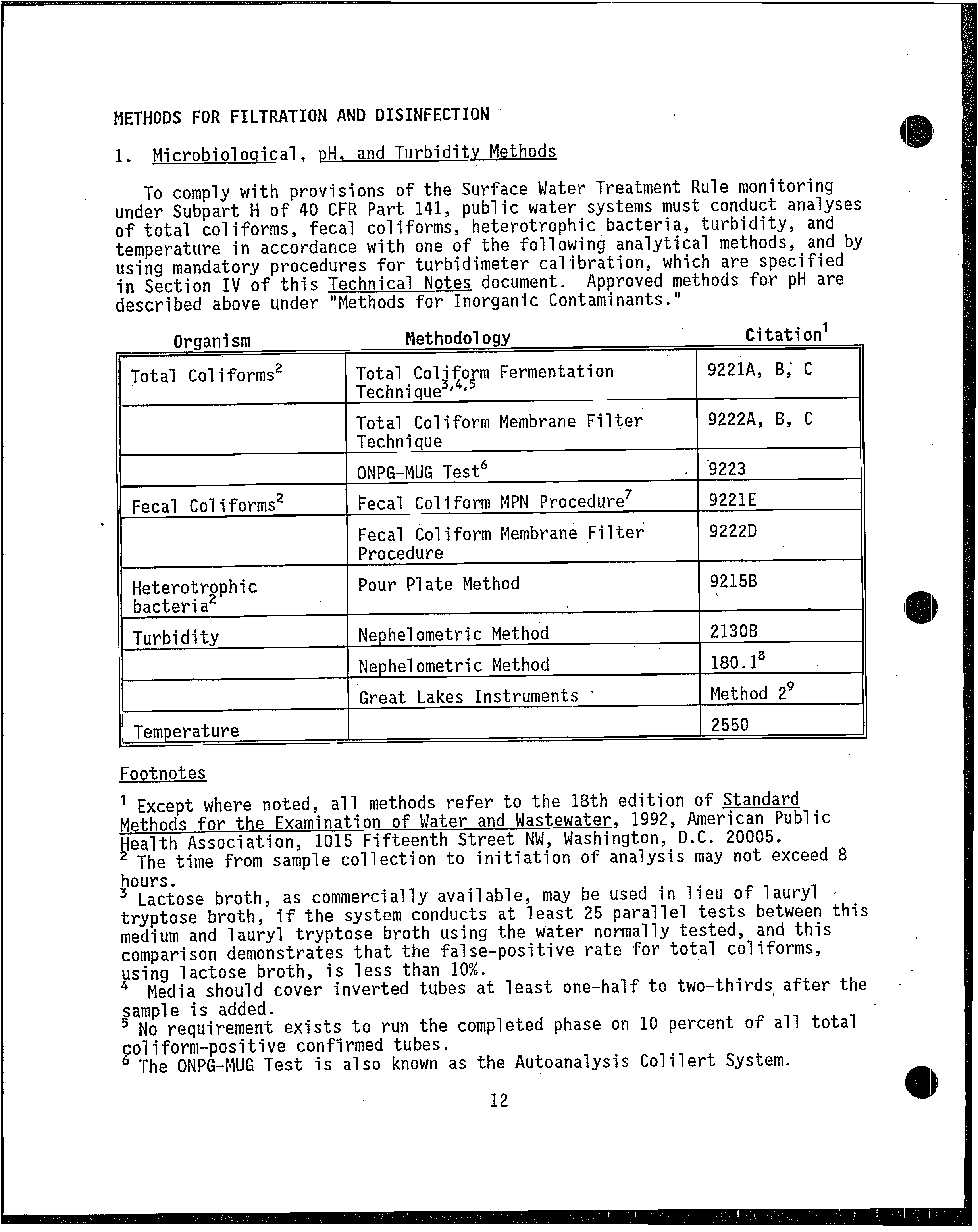
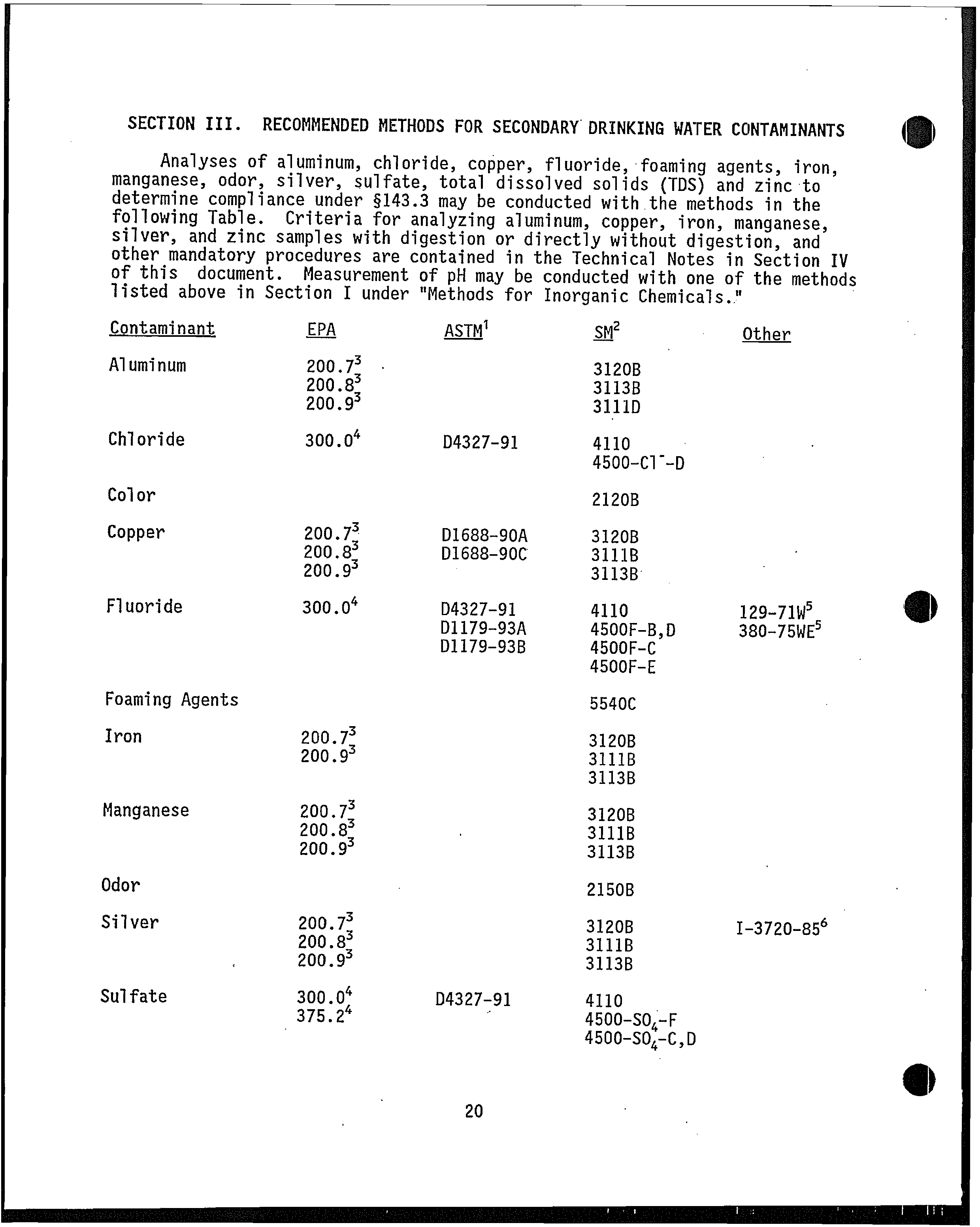
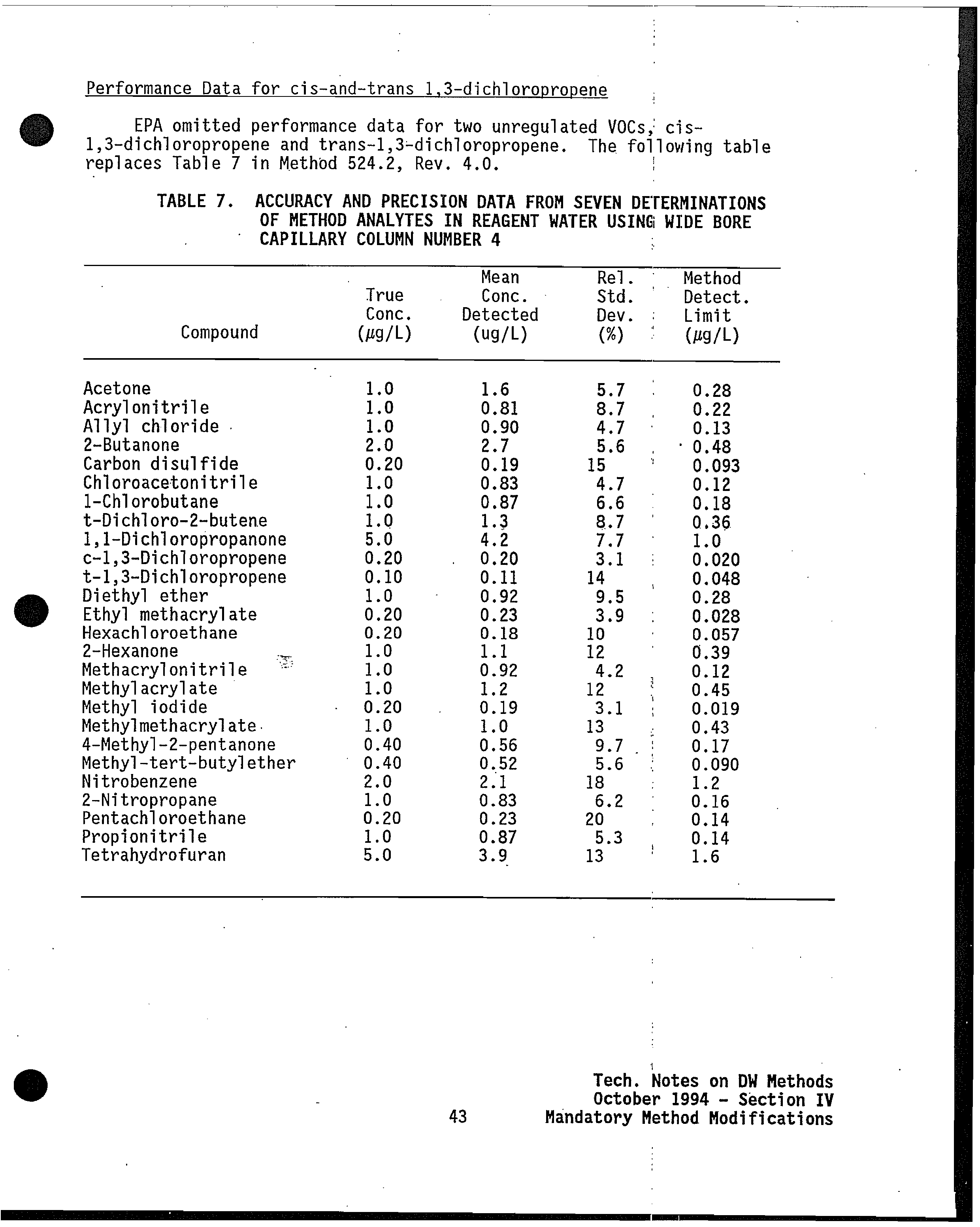
From:
<Fair.Pat©epamail.epa.gov>
A L.
Date:
6/30/2008 12A8:07 PM
Subject:
here
it is
RECEUVED
CLERK’S
OFFICE
see
page 14 (24th
page of
the
file)
NOV
262008
(See attached file:
Tech Notes.pdf)
STATE
OF
Pollution
Control
8or.d
7)6V)7/1
/fS
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
6/30/2008
5:37:42
PM
Subject:
Re: here it is
Thank
you for forwarding the reference.
I have checked the lists of
discontinued methods,
those
recommended
for further
use, and those still in
use. I have noted two apparent discrepancies
that I hope you can
clarify.
First,
page 15 of Technical Notes indicates
that Method 245.1 was discontinued
for mercury, yet page 5 lists Method
245.1
from
Supplement las recommended
for mercury, and 40 C.F.R. 141 .23(k)(1)
lists this method for Mercury. Is this
correct?
Was
the shift from
a
prior version
to that in Supplement I?
Second,
page
18
lists Method 502.2 as discontinued
for 1,2-dichlorobenzene, yet
page 8 lists it for use and 40 C.F.R.
141 .24(e)(1)
also
lists
it for 1,2-dichlorobenzene.
Is
the
appearance on page 18 of Technical
Notes a typographic error
that
should
have
listed Method 502.1 as discontinued?
Please clarify these
issues to me if you are
able
to
do so. Alternatively, let me know if
you cannot do so at this time.
Michael
J.
McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Pat©epamail.epa.gov>
6/30/2008 12:42 PM >>>
see page 14 (24th page of the file)
(See attached file: Tech Notes.pdf)
From:
Mike McCambridge
To:
Fair.PatepamaiI.epa.gov
Date:
7/1/2008 11:10:39AM
Subject:
Re: here it is
Thanks.
No great rush.
Michael J. McCambridge
Attorney
Illinois Pollution
Control Board
312-814-6924
>>> <Fair.Patepamail.epa.gov>
7/1/2008 6:24 AM >>>
Mike,
I’ll check into
your
questions,
but I can’t guarantee that I’ll
resolve
them.
There isn’t any one here who was involved
in putting Technical
Notes together,
so
I will
need to do some research. I’ll let
you
know
what
I find out; it may be next week before I
get back to you.
Pat
“Mike
McCambridge”
<mccambridge©ipc
To
b.state.il.us>
Pat
Fair/Cl/USEPA/US@EPA
cc
06/30/2008
06:37
PM
Subject
Re: here it is
Thank you for forwarding the reference. I have checked
the lists of
discontinued methods,
those
recommended for further use, and those still
in use. I have noted two apparent discrepancies that I hope
you can
clarify.
First,
page
15 of Technical Notes indicates that Method
245.1
was
discontinued for mercury, yet page 5 lists Method 245.1 from
Supplement
I
as
recommended for mercury, and 40 C.F.R. 141
.23(k)(1) lists this
method for Mercury. Is this correct? Was the shift from
a
prior
version to that in Supplement I?
Second, page 18 lists
Method 502.2
as
discontinued
for
1,2-dichlorobenzene, yet page 8 lists it for
use
and 40
C.F.R. 141.24
(e)(1) also lists it for 1 ,2-dichlorobenzene. Is the appearance
on page
18 of Technical Notes a
typographic
error
that
should have listed Method
502.1
as
discontinued?
Please
clarify these issues to me if
you
are able to
do so.
Alternatively, let me know if you cannot do so at this time.
Michael J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Patepamail.ega.Qov>
6/30/2008
12:42 PM >>>
see
page
14 (24th
page of the file)
(See attached file: Tech Notes.pdf)
From:
<Fair.
Pat@epamail.epa.gov>
To:
MCCAMBM©ipcb.state.iI.us
Date:
7/1/2008
2:35:08
PM
Subject:
Re: here
it is
Mike,
I have found the answers
to your
questions:
1. Prior
to
the Dec 5,1994
rule, the citation
for EPA
Method 245.1
(issued
in 1974) was “Methods
of Chemical
Analysis
of Water
and Wastes,”
EPA Environmental
and
Monitoring
and
Support
Laboratory,
Cincinnati, OH
45268
(EPA-600/4-79-020).
March 1983.
Availablefrom
ORD Publications,
CERI, EPA,
Cincinnati,
OH 45268.
The
method was
updated to the version
(Revision
3.0) that is published
in “Methods
for Determination
of Metals
in Environmental
Samples,
Supplement
I” (EPA-600/R-94-1
11) May 1994
which
was approved in the Dec
5, 1994 rule.
2.
A
similar situation exists for
EPA
502.2.
The original
citation
was
“Methods
for the Determination
of Organic
Compounds in Drinking
Water,”
ORD
Publications, CERI,
EPAI600I4-881039,
December
1988. The manual was
revised in
July 1991
and
the
methods in the
revised manual were
approved
in the
Dec 5, 1994 rule. Method
502.2, Revision
2.0
(1989)
replaced
Revision
1.0 (1986).
Also
note
that
there has
been another
change to EPA
502.2. The version
that is
now
cited is
Revision 2.1 (1995)
which is published
in “Methods
for
the Determination
of Organic Compounds
in Drinking
Water, Supplement
Ill” EPAI600/R-95-131,
August
1995.
The previous version
was withdrawn
effective
June
1, 2001.
Hope
this helps.
Pat
“Mike
McCambridge”
<mccambridge©ipc
To
b.state.il.us>
Pat
Fair/CI/USEPAIUS@EPA
cc
06/30/2008
06:37
PM
Subject
Re: here it
is
Thank
you for forwarding the
reference.
I have
checked the
lists of
discontinued methods,
those recommended for further
use, and those still
in use. I have noted
two apparent discrepancies that
I hope you can
clarify.
First,
page 15 of Technical Notes indicates that
Method 245.1 was
discontinued
for mercury, yet page
5
lists Method 245.1
from Supplement
las recommended
for mercury, and 40 C.F.R. 141 .23(k)(1)lists
this
method for Mercury.
Is
this
correct? Was the shift from
a
prior
version to that in Supplement I?
Second,
page 18 lists Method 502.2
as
discontinued
for
I ,2-dichlorobenzene, yet
page 8 lists it for use and 40 C.F.R. 141.24
(e)(1) also lists it for 1 ,2-dichlorobenzene. Is the
appearance on page
18 of Technical Notes
a typographic error that should have listed Method
502.1 as discontinued?
Please
clarify these issues to me if
you
are able to
do so.
Alternatively, let me know if
you cannot do so at this time.
Michael J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Pat©epamail.epa.gov> 6/30/2008 12:42
PM >>>
see page
14 (24th
page
of the
file)
(See attached file: Tech Notes.pdf)
From:
Mike McCam bridge
To:
Fair.Pat©epamail.epa.gov
Date:
7/1/2008 2:37:33
PM
Subject:
Re: here it is
Thank
you. I will use
this insight in my review to assure that the Illinois rules comport with
the
minimum
federal
requirements.
Michael
J. McCambridge
Attorney
Illinois Pollution
Control Board
312-814-6924
>>> <Fair.Patepamail.epa.gov>
7/1/2008 2:34
PM >>>
Mike,
I have
found
the
answers to your questions:
1. Prior to the
Dec 5, 1994 rule, the citation for EPA Method
245.1
(issued in 1974) was “Methods
of Chemical Analysis of Water and Wastes,”
EPA Environmental
and Monitoring and Support Laboratory, Cincinnati,
OH
45268 (EPA-600/4-79-020). March 1983.
Available from ORD Publications,
CERI, EPA, Cincinnati,
OH 45268. The method was updated to the version
(Revision 3.0) that is published in “Methods
for Determination of Metals
in Environmental
Samples, Supplement I” (EPA-600/R-94-111) May 1994
which was approved in the Dec
5,
1994
rule.
2. A similar situation exists for EPA
502.2. The original citation was
“Methods for the Determination of Organic
Compounds
in Drinking Water,”
ORD Publications,
CERI, EPA’600/4-88/039, December 1988. The manual was
revised in July 1991 and the methods
in
the
revised manual were approved
in the Dec 5, 1994 rule. Method 502.2, Revision 2.0 (1989)
replaced
Revision 1.0(1986).
Also note that there has been another change to EPA 502.2.
The version
that
is now
cited
is Revision 2.1 (1995) which is published in “Methods
for the Determination of Organic Compounds
in
Drinking Water,
Supplement
Ill” EPA/600/R-95-131, August 1995. The previous version was withdrawn
effective
June
1, 2001.
Hope
this
helps.
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
06/30/2008
06:37
PM
Subject
Re: here it is
Thank you
for
forwarding the reference. I have checked the
lists of
discontinued methods,
those
recommended for further use, and those still
in use.
I have noted two apparent discrepancies that I hope
you can
clarify.
First,
page 15 of
Technical Notes
indicates that
Method 245.1
was
discontinued
for mercury,
yet page 5
lists
Method 245.1
from Supplement
I as
recommended
for
mercury, and
40 C.F.R. 141
.23(k)(1)
lists
this
method for
Mercury.
Is
this
correct? Was
the shift
from a prior
version
to that in
Supplement
I?
Second,
page
18 lists
Method 502.2
as
discontinued
for
1,2-dichlorobenzene,
yet
page 8 lists it
for
use
and
40 C.F.R.
141.24
(e)(1)
also lists
it for 1 ,2-dichlorobenzene.
Is
the appearance
on
page
18 of Technical
Notes
a
typographic
error that should
have listed
Method
502.1
as
discontinued?
Please
clarify these
issues to
me if
you are able
to do so.
Alternatively,
let
me know
if
you cannot
do so
at this
time.
Michael
J.
McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
>>> <Fair.Pat(äeoamail.ea.gov>
6/30/2008
12:42 PM >>>
see
page 14 (24th
page of the
file)
(See
attached
file:
Tech Notespdf)
From:
<Fair.
Pat©epamail.epa.gov>
To:
MCCAMBM@ipcb.state.il.us
Date:
7/1/2008
2:40:12 PM
Subject:
Re:
here it is
Are
you adding references
to the new appendix that
includes optional
alternative methods?
Just
curious...
“Mike
McCam bridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
07/01/2008 03:37
PM
Subject
Re:
here it is
Thank you. I will use this insight in
my review to assure that the
Illinois rules
comport with the minimum federal
requirements.
Michael J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Pat©epamail.epa.gov>
7/1/2008 2:34 PM >>>
Mike,
have
found the answers
to your questions:
1. Prior
to the Dec
5,
1994
rule, the citation for EPA Method
245.1
(issued in 1974) was “Methods of
Chemical Analysis of Water and Wastes,”
EPA
Environmental
and Monitoring and Support Laboratory,
Cincinnati, OH
45268 (EPA-600/4-79-020).
March 1983. Available from ORD
Publications,
CERI, EPA, Cincinnati, OH 45268. The
method
was updated to the version
(Revision 3.0)
that
is published in “Methods for Determination
of Metals
in
Environmental Samples,
Supplement I” (EPA-600/R-94-1
11) May 1994
which was approved in the Dec
5,
1994
rule.
2. A
similar situation exists for
EPA 502.2. The original citation
was
“Methods
for the Determination
of Organic
Compounds in Drinking Water,”
ORD Publications, CERI, EPA/600/4-88/039,
December 1988.
The manual was
revised
in July 1991 and the
methods in the revised
manual were approved
in the Dec 5, 1994 rule. Method 502.2,
Revision 2.0 (1989) replaced
Revision
1.0
(1986).
Also
note that there
has been another
change to
EPA
502.2. The version
that
is now cited is
Revision
2.1 (1995)
which is published
in “Methods
for the
Determination
of Organic
Compounds in Drinking
Water, Supplement
Ill’ EPA1600/R-95-131,
August 1995.
The previous
version
was
withdrawn
effective June
1, 2001.
Hope
this helps.
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPA/US@EPA
cc
06/30/2008
06:37
PM
Subject
Re:
here it is
Thank
you for
forwarding
the reference. I have
checked the lists
of
discontinued
methods, those
recommended
for further use, and those
still
in use. I have noted
two
apparent discrepancies
that
I hope you
can
clarify.
First,
page 15 of Technical Notes
indicates that
Method 245.1 was
discontinued
for mercury, yet
page 5 lists Method
245.1 from
Supplement
I as recommended
for mercury,
and 40 C.F.R.
141 .23(k)(1)lists this
method for
Mercury. Is this correct?
Was the shift
from
a
prior
version
to that in Supplement
I?
Second,
page 18 lists Method
502.2 as discontinued
for
1
,2-dichlorobenzene,
yet page
8 lists it for use and
40 C.F.R. 141.24
(e)(1)
also lists it for 1 ,2-dichlorobenzene.
Is the
appearance on page
18 of Technical
Notes
a
typographic
error that should
have listed
Method
502.1 as
discontinued?
Please clarify these
issues to me if you are
able to
do so.
Alternatively,
let
me
know
if
you
cannot
do so at this time.
Michael
J.
McCambridge
Attorney
Illinois
Pollution Control
Board
312-814-6924
>>> <Fair.PatepamaiI.epa.gov>
6/30/2008
12:42 PM >>>
see page 14 (24th
page of the file)
(See attached
file: Tech Notes.pdf)
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
7/1/2008
2:59:32
PM
Subject:
Re: here
it is
I am uncertain
how best to
deal
with
the appendix
listing of alternative
methods.
It is possible
that
the listing itself
may
be
added to the Illinois rules
as an appendix.
It is also
possible
that
references in the
various federally
derived provisions
that
restrict
the
selection
of methods
(i.e., the State
counterparts to 40 C.F.R.
141.23(e)(1),
141.24(k)(1),
etc.) will
require a
reference to
the
listing of alternative methods.
I should have
a clearer
picture
as I continue my work
on
the
proposal, after
I
have
dealt
with the USEPA March
12, 2007 amendments.
Michael J.
McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
>>> <Fair.Pat©epamail.epa.gov>
7/1/2008 2:39
PM >>>
Are
you
adding references
to the new appendix
that
includes
optional
alternative
methods? Just curious...
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
07/01/2008 03:37
PM
Subject
Re: here
it is
Thank
you.
I will
use this
insight
in my review to assure that
the
Illinois rules comport with
the minimum federal requirements.
Michael
J. McCambridge
Attorney
Illinois
Pollution Control Board
312-814-6924
>>> <Fair.Pattä.eamaiI.epa.gov>
7/1/2008
2:34 PM
>>>
Mike,
I have found the answers
to your questions:
1.
Prior to the Dec 5, 1994 rule,
the citation for EPA Method
245.1
(issued in 1974)
was
“Methods
of
Chemical
Analysis of Water and Wastes,”
EPA Environmental
and Monitoring
and Support
Laboratory,
Cincinnati, OH
45268 (EPA-600/4-79-020).
March 1983. Available
from
ORD
Publications,
CERI,
EPA,
Cincinnati, OH 45268.
The method was updated
to the version
(Revision 3.0)
that is published
in “Methods
for Determination of Metals
in Environmental
Samples, Supplement
I” (EPA-600/R-94-1
11) May
1994
which was
approved in the Dec
5, 1994 rule.
2. A similar
situation exists for EPA
502.2.
The original
citation was
“Methods for the
Determination
of
Organic Compounds
in Drinking
Water,”
ORD
Publications,
CERI, EPAI600/4-88/039,
December 1988.
The manual was
revised in July 1991 and
the methods
in the
revised manual
were
approved
in the Dec
5, 1994 rule. Method 502.2,
Revision 2.0 (1989) replaced
Revision 1.0 (1986).
Also note that
there
has been another
change to EPA 502.2. The version
that is now
cited
is Revision 2.1 (1995) which
is published in “Methods
for the Determination
of
Organic
Compounds in Drinking
Water, Supplement
Ill”
EPAJ600/R-95-131, August
1995. The previous version
was
withdrawn
effective
June 1,2001.
Hope this helps.
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/CI/USEPNUS@EPA
cc
06/30/2008 06:37
PM
Subject
Re: here it is
Thank you for forwarding the reference.
I
have
checked
the lists of
discontinued
methods, those recommended for further
use, and those still
in
use. I
have noted two apparent
discrepancies that I hope
you
can
clarify.
First,
page
15
of
Technical Notes indicates
that
Method 245.1 was
discontinued for mercury, yet page 5 lists Method 245.1
from Supplement
I as recommended for
mercury,
and 40 C.F.R. 141 .23(k)(1) lists this
method for Mercury. Is this correct? Was the shift
from a prior
version to that in
Supplement
I?
Second, page 18 lists
Method
502.2 as discontinued for
1 ,2-dichlorobenzene, yet page 8 lists it for
use and 40 C.F.R. 141.24
(e)(1) also
lists
it for 1
,2-dichlorobenzene.
Is the appearance on
page
18 of Technical Notes a typographic error that should have
listed Method
502.1 as
discontinued?
Please
clarify these issues to me if
you are able to do so.
Alternatively, let me know if you cannot do so at this time.
Michael
J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Patepamail.epa.gov>
6/30/2008 12:42 PM >>>
see page
14(24th
page of the file)
(See attached file:
Tech Notes.pdf)
From:
Mike
McCambrdge
To:
Fair.Pat©epamail.epa.gov
Date:
7/8/2008
12:57:19
PM
Subject:
Waters
Methods
I have tried
to obtain
copies
of the two Waters
methods
referenced
in 40 C.F.R.
141.23(k)(1)
for fluoride
and nitrite/nitrate
using
the contact
information
included
in the
rule. At first,
the Waters
rep could
not
locate
anything
based
on
the
EPA
descriptions
included
in the
rule. This
morning I received
two documents
that
purport to
be the
methods.
The documents
raise
questions
that you
might
answer
for
me.
The
copy of Method
B-i
011
sent
me by Waters
is nearly
identical
to one
that
found
on
the USEPA
website.
The only
difference
between the
two is that the
method from
the USEPA website
is headed
“Waters.”
The document
it appears
to
include
pages 13
through 17 from
some reference.
It is undated,
which
means
that
I cannot
use it for
an
incorporation
by
reference. Do
you
have
a
dated copy of
Method B-i 011
or
a
fuller copy
of
the
posted
reference
that would
include the
date?
It appears
that the method
is just one
cited out of
a
fuller reference,
and
I should cite
to that
fuller reference
by its
own title.
I
will
also approach
Waters
with
this request.
Your
rule cites “Waters
Method
D6508, Rev. 2,”
entitled “Test
Method for
Determination
of
Dissolved
Inorganic
Anions in
Aqueous Matrices
Using
Capillary
Ion Electrophoresis
and
Chromate
Electrolyte.”
Waters sent
me
a document
marked
“Method
6500,” “revision
0,”
and dated
February
2007,” and entitled
“Dissolved
Inorganic
Anions
In Aqueous
Matrices
By
Capillary
Ion Electrophoresis.”
That document
appears to
be Method 6500
from SW-846.
Is
“Waters
Method D6508,
Rev.
2”
the same
as
Method
6500,
rev.
0 from
SW-846?
If
so,
why did
USEPA cite
this
as
“D6508”?
If not, can
you
forward
me
a copy
of Method
D6508
or
give
me enough information
to identify
the method
to Waters,
that I might
obtain
a
copy of the
right method?
Michael
J. McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
Page 1
of2
Mike
McCambridge
- Re: Waters
Methods
From:
<Fair.Patepamail.epa.gov>
To:
“Mike
McCambridge”
<mccambridgeipcb.state.il.us>
Date:
7/8/2008
2:19 PM
Subject:
Re: Waters
Methods
Mike,
I’m
working
off
site
today,
so
I
don’t have
access to
the references
I need
to
answer
your
questions.
I should
have copies
of the
methods
that were
added to
40 CFR
141
as part of
the 2007
methods
update
rule. If these
Waters
methods
are prior
to that,
I might
not be
able
to help
you.
Unfortunately,
I don’t
know
who
might
have
them
other
than Waters.
I’ll see what
I can find
tomorrow
and
get back
to
you.
Pat
11
Mike
McCambridge”
<mccambridge@ipcb.state.iLus>
wrote:
To: Pat
Fair/CI/USEPA/US@EPA
From:
“Mike McCambridge”
<mccambridge@ipcb.state.il.us>
Date:
07/08/2008
01:57PM
Subject:
Waters
Methods
I have
tried to
obtain
copies
of
the two
Waters
methods
referenced
in
40 C.F.R.
141.23(k)
(1)
for
fluoride
and
nitrite/nitrate
using
the
contact
information
included
in
the
rule.
At
first,
the
Waters
rep
could
not locate
anything
based
on
the EPA
descriptions
included
in
the
rule.
This
morning
I
received
two
documents
that
purport
to
be
the
methods.
The
documents
raise
questions
that
you
might
answer
for
me.
The
copy of
Method
B-lOll
sent
me
by Waters
is nearly
identical
to
one
that
I
found
on the
USEPA
website.
The
only difference
between
the
two
is that
the
method
from
the
[JSEPA
website
is headed
“Waters.”
The
document
it
appears
to
include
pages
13 through
17
from some
reference.
It is
undated,
which
means that
I cannot
use
it
for an
incorporation
by
reference.
Do
you
have
a dated
copy
of
Method
B-lOll
or a fuller
copy
of
the
posted
reference
that
would
include
the
date?
It
appears
that
the method
is just
one
cited
out
of a
fuller
reference,
and I should
cite
to
that fuller
reference
by
its
own title.
I will
also
approach
Waters
with
this
request.
Your
rule cites
“Waters
Method
D6508,
Rev.
2,”
entitled
“Test
Method
for
Determination
of Dissolved
Inorganic
Anions
in Aqueous
Matrices
Using
Capillary
Ion Electrophoresis
and
Chromate
Electrolyte.”
Waters
sent
me
a document
marked
“Method
6500,”
“revision
0,”
and
dated
February
2007,”
and
entitled
“Dissolved
Inorganic
Anions
In
Aqueous
Matrices
By
Capillary
Ion
Electrophoresis.”
That
document
file
://C :\Documents
and Settings\McCambM\Local
Settings\Temp\GW}
00002.HTM
11/26/2008
Page 2
of2
appears
to be
Method
6500
from
SW-846.
Is
T
Waters
Method
1D6508,
Rev.
2T
the same
as Method
6500,
rev.
0 from
SW-846?
If
so, why
did
USEPA cite
this as
“D6508’
T
?
If not,
can
you
forward
me a copy
of
Method D6508
or give
me enough
information
to identify
the method
to
Waters,
that
I might
obtain a
copy of the
right method?
Michael
J. McCambridge
Attorney
Illinois
Pollution
Control Board
312-814-6924
file://C
:\Documents
and Settings\McCambM\Local
Settings\Temp\GW} 00002.HTM
11/26/2008
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
7/8/2008
2:29:21 PM
Subject:
Re: Waters
Methods
Whatever
you can do for me
when you get back.
I have continued to
look into this today.
I am convinced that Method
6500 added
to SW-846
in Update
IV in the end
of 2007 is the method
you have called “Method
D6508”
from
Waters.
See 73 Fed.
Reg. 486
(Jan.
3,
2008);
http://v,ww.eoa.govISW-846/rjdfs/6500.pdf.
If this
is
true,
I will likely cite the SW-846
version
of
the method,
since
it
is much easier to obtain
than
the method
from Waters. As
described,
Waters
initially acted like I spoke
a foreign language
when
I
asked
for ‘Method D6508.”
As for Method B-i
Oil,
it
seems to distill down
to
me needing
the
title to the document in
which
the
method
appears.
Talk to you when you return.
Michael J. McCambridge
Attorney
Illinois
Pollution Control Board
312-814-6924
>>> <Fair.Pat1epamaiI.epa.gov>
7/8/2008
2:18 PM >>>
Mike,
I’m working off site today,
so I don’t have
access to the references
I need to answer your
questions.
I should
have copies
of the
methods that were
added to 40 CFR
141
as part of the
2007 methods update
rule. If these Waters
methods
are
prior
to
that, I
might not be able to help
you. Unfortunately, I
don’t know who
might
have them other than Waters.
I’ll see
what I can find tomorrow
and
get back to
you.
Pat
“Mike McCambridge”
<mccambrideCtipcb.state.il.us>
wrote:
To:
Pat
Fair/CI/USEPA/US@EPA
From: “Mike McCambridge”
<mccambrideäicb.state.iI.us>
Date: 07/08/2008 01 :57PM
Subject: Waters
Methods
I have tried to obtain
copies of the two Waters
methods referenced
in 40
C.F.R. 141
.23(k)(i) for fluoride
and
nitrite/nitrate
using
the
contact information
included
in the rule. At first, the Waters
rep could not locate
anything
based
on the EPA descriptions
included
in the rule. This morning
I
received two
documents
that purport to
be
the methods.
The
documents raise questions
that you
might
answer for
me.
The copy
of Method
B-loll sent me by Waters
is nearly identical
to one that I found on
the
USEPA website.
The only
difference
between
the two is that the method
from the
USEPA
website is headed “Waters.”
The document it
appears to
include
pages
13
through 17 from
some
reference. It is undated,
which means that
I cannot use it for an incorporation
by reference.
Do
you have
a
dated copy
of Method B-loll or a fuller
copy
of the posted
reference that would include
the date? It appears
that the
method
is
just one
cited out of
a
fuller
reference, and I should cite
to
that fuller
reference
by its own title. I will
also approach Waters
with
this
request.
Your rule
cites
“Waters
Method D6508, Rev. 2,” entitled
“Test Method
for Determination
of
Dissolved
Inorganic Anions
in Aqueous
Matrices Using Capillary
Ion Electrophoresis
and Chromate Electrolyte.”
Waters sent
me a document
marked “Method 6500,”
“revision
0,” and dated February
2007,” and entitled “Dissolved
Inorganic Anions
In Aqueous
Matrices By Capillary Ion
Electrophoresis.” That
document appears
to be Method 6500 from
SW-846. Is “Waters
Method D6508,
Rev. 2” the same
as
Method
6500, rev. 0 from SW-846?
If
so,
why did
USEPA
cite this as “D6508”? If
not,
can you forward
me
a copy of
Method
D6508 or
give
me
enough information to identify
the method to Waters,
that I might obtain
a copy of the right method?
Michael J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
From:
<Fair.Pat©epamail.epa.gov>
To:
MCCAMBM©ipcb.state.iI.us
Date:
7/10/2008
7:46:26
AM
Subject:
Re:
Waters
Methods
Mike,
Here’s
Waters
6508
method
that
was
approved
for drinking water. Aren’t
you
limited to
methods that
are
listed as
approved
for drinking water?
Based
on the info at
the top of the
method,
I’m
guessing this
may
now be
an ASTM
method.
t was
evaluated
under
the ATP program,
so
EPA
was
given the method
prior to the
ASTM process.
I don’t know this
for sure
and it will be something
I investigate
as we
begin putting together
our
next Expedited
Methods Approval
FR action.
(If it is the same
method,
we’ll probably
list it
in Appendix
A.)
(See attached
file: Waters
Method
D
6508,
Rev
2_EPA-HQ-OW-2003-0070-0063
.pdf)
As for the
other method, it was
approved prior to
the
2007 methods
rule.
I don’t have
a
copy
of
it,
because
I wasn’t
involved
in the earlier
methods
rules. However, I have
asked our ATP
coordinator to
see if it
is in the ATP file.
When
I hear
back from
him,
I’ll let you know.
Hope this helps,
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
07/08/2008
03:29
PM
Subject
Re: Waters Methods
Whatever
you can do for me when
you
get
back.
I have continued to
look
into
this today.
I am convinced
that Method
6500 added to SW-846
in
Update IV
in
the end of 2007 is the
method
you
have called “Method
D6508”
from
Waters. See 73
Fed. Reg. 486
(Jan. 3, 2008);
http://www.epa.gov/SW-846/pdfs/6500.pdf.
If this
is true,
I will
likely
cite the
SW-846
version
of
the
method,
since it
is much easier to obtain
than the method
from Waters. As
described, Waters
initially acted
like
I
spoke
a
foreign
language when
I asked for “Method
D6508.”
As for
Method
B-i Dli, it
seems to distill down
to me needing
the title to
the
document
in which
the
method appears.
Talk to
you
when
you return.
Michael
J.
McCam
bridge
Attorney
Illinois
Pollution Control
Board
312-814-6924
>>> <Fair.Pat©epamail.epa.gov>
7/8/2008
2:18
PM >>>
Mike,
I’m
working off
site today,
so
I don’t have
access
to the references
I
need to answer
your questions.
I
should have copies
of the methods that
were
added to
40 CFR 141
as
part of the 2007
methods update rule.
If
these Waters methods
are prior to
that,
I might
not be able to help
you.
Unfortunately,
I don’t know who
might have them other
than
Waters.
I’ll
see what I can
find tomorrow
and get back to
you.
Pat
“Mike McCambridge”
<mccambridge©ipcb.state.il.us>
wrote:
To: Pat
Fair/Cl/USEPAIUS@EPA
From:
“Mike McCambridge”
<mccambridgeipcb.state.il.us>
Date: 07/08/2008
01 :57PM
Subject:
Waters Methods
I have
tried to obtain
copies of the two Waters
methods referenced
in 40
C.F.R. 141 .23(k)(1) for fluoride
and
nitrite/nitrate
using
the
contact
information
included
in the rule. At first, the
Waters
rep could
not
locate anything based on
the EPA
descriptions
included
in the rule.
This morning I received
two
documents that
purport to be
the methods.
The
documents raise questions
that
you
might answer for
me.
The
copy of Method B-i
011 sent me
by
Waters is nearly identical
to
one
that I
found on the USEPA website.
The only
difference
between the two
is that the method from
the USEPA website
is headed “Waters.”
The
document
it appears to include
pages 13 through
17
from
some reference.
It is
undated,
which means
that I cannot
use
it
for an incorporation
by
reference.
Do
you
have a dated
copy of Method
B-loll
or a fuller copy
of
the posted reference
that would include the
date? It appears
that
the
method
is just
one
cited out
of a fuller reference,
and
I should
cite
to that fuller reference
by its own
title.
I will also approach
Waters with this request.
Your rule cites “Waters
Method D6508,
Rev. 2,” entitled
“Test Method for
Determination
of Dissolved Inorganic Anions
in Aqueous
Matrices
Using
Capillary
Ion Electrophoresis
and
Chromate Electrolyte.”
Waters sent me
a
document marked
“Method 6500,” “revision
0,” and
dated
February
2007,”
and
entitled “Dissolved
Inorganic
Anions
In Aqueous
Matrices By
Capillary Ion Electrophoresis.”
That
document appears
to
be
Method
6500
from
SW-846. Is “Waters
Method
D6508,
Rev. 2” the
same as Method
6500,
rev. 0 from
SW-846?
If so, why
did USEPA
cite
this
as “D6508”?
If not,
can
you
forward me
a
copy of
Method
D6508
or
give me enough
information
to identify
the
method
to Waters,
that
I might obtain
a copy
of the
right method?
Michael
J. McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
7/10/2008
5:07:33 PM
Subject:
Re: Waters Methods
Thank you. That
nails it down.
I
will cite it
as an ASTM
method.
I have
another
method problem. I
have been trying to obtain
a copy of that
Ra-226/Ra-228 method
by gamma-ray
spectometry developed
by Georgia Insitute of
Technology. The
“Environmental Resources
Center” has been disbanned
or
something,
so
that
the
number at
40 C.F.R.
141.74
is no longer valid. It may
have
become
the
Environmental
Radiation
Center
or
something.
I have placed several
calls and e-mails with
Bernd Kahn
and
the
Center in an attmpt to locate
the
method,
but
no luck so far.
Can you help on
this one too?
Michael J. McCambridge
Attorney
Illinois
Pollution Control Board
312-814-6924
>>> <Fair.Pat@epamail.epa.gov>
7/10/2008
7:43 AM >>>
Mike,
Here’s Waters
6508 method that was approved
for drinking
water. Aren’t
you
limited
to methods that
are
listed
as approved for
drinking
water?
Based on the info
at the top of the method,
I’m guessing this may
now be
an ASTM
method. It was
evaluated
under the ATP
program, so EPA was
given the method prior to
the ASTM process.
I don’t know this for sure
and it
will be something I investigate
as we
begin
putting
together our
next Expedited Methods Approval
FR action. (If
it is the
same
method,
we’ll probably
list it in Appendix A.)
(See
attached
file:
Waters Method
D 6508, Rev
2_EPA-HQ-OW-2003-0070-0063.pdf)
As for
the other method, it was approved
prior to the
2007 methods rule.
I don’t have
a
copy
of it, because I wasn’t involved
in the earlier
methods rules.
However,
I have
asked our ATP coordinator
to
see if it
is in the ATP file. When
I hear back from him,
I’ll let you know.
Hope this
helps,
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
07/08/2008
03:29
PM
Subject
Re:
Waters
Methods
Whatever you
can do for me when
you get back. I
have continued to look
into this today.
I am
convinced
that Method
6500 added to SW-846 in
Update
IV in the end of 2007 is
the
method
you have
called “Method
D6508” from Waters.
See
73
Fed. Reg. 486
(Jan.
3,
2008):
http://www.epa.gov/SW-846/pdfs/6500.pdf.
If this is true, I will likely
cite the SW-846 version
of the method, since it
is much easier to obtain
than the method from Waters.
As described, Waters initially acted
like
I
spoke a foreign language when
I asked for “Method D6508.” As for
Method
B-lOll, it seems to distill down to
me needing the title to the
document
in
which the method
appears.
Talk to you when you return.
Michael
J. McCambridge
Attorney
Illinois
Pollution Control Board
312-814-6924
>>> <Fair.Pategamail.ega.gov>
7/8/2008
2:18 PM >>>
Mike,
I’m working
off
site today,
so I don’t have access to the references
I
need to answer your questions. I should
have copies of the methods that
were
added
to 40 CFR 141
as part of the 2007 methods update
rule. If
these
Waters methods are prior to that, I
might not be able to help you.
Unfortunately, I don’t know who
might have them other than Waters.
I’ll see what I can find tomorrow and
get back to you.
Pat
“Mike McCambridge’
<mccambridoeipcb.state.il.us> wrote:
To: Pat
Fair/CI/USEPNUS@EPA
From: “Mike McCambridge” <mccambridgeipcb.state.il.us>
Date:
07/08/2008 01 :57PM
Subject: Waters Methods
I have tried to obtain copies of the two Waters methods referenced
in 40
C.F.R.
141 .23(k)(1)for fluoride
and nitrite/nitrate using the contact
information included in the rule. At first, the Waters
rep could not
locate anything
based on
the
EPA descriptions included in the rule.
This morning I received two documents that
purport to be the methods.
The documents raise questions that you might answer for me.
The copy
of Method B-i 011 sent me by Waters is nearly identical to
one
that
I found on the USEPA website. The only
difference between the two
is that the method from the USEPA website is headed “Waters.” The
document it appears to include pages 13 through
17 from some reference.
It is
undated, which
means
that
I cannot use it for an incorporation
by
reference. Do you have a dated copy of Method B-i 011
or a fuller copy
of the
posted reference that would
include the date? It appears that
the method is just one cited out of a fuller reference, and
I should
cite
to that fuller reference
by
its own title.
I will also approach
Waters
with this request.
Your rule cites “Waters Method D6508, Rev. 2,” entitled “Test
Method for
Determination of
Dissolved
Inorganic
Anions in Aqueous Matrices Using
Capillary Ion Electrophoresis and Chromate Electrolyte.” Waters
sent me
a
document marked “Method 6500,” “revision 0,’ and dated February
2007,”
and
entitled “Dissolved Inorganic Anions
In Aqueous Matrices By
Capillary Ion Electrophoresis.” That document appears to
be Method 6500
from SW-846. Is “Waters
Method
D6508, Rev. 2” the same as Method
6500,
rev. 0 from
SW-846?
If
so, why did USEPA cite this
as ‘D6508”? If not,
can you forward me
a
copy of Method D6508 or give me
enough information
to identify the method to
Waters, that
I might obtain a copy of the
right method?
Michael
J.
McCambridge
Attorney
Illinois Pollution Control
Board
312-814-6924
Page 1 of5
Mike
McCambridge
- Re:
Waters
Methods
From:
<Fair.Patepamai1.epa.gov>
To:
“Mike
McCambridge”
<mccambridgeipcb.state.i1.us>
Date:
7/10/2008
9:31
PM
Subject:
Re: Waters
Methods
Mike,
I
haven’t
done
a one-to-one
check
of
the
ASTM
method
against
the
Waters
method,
so I
can’t say
for
sure that
they are
the
same.
My
comment was
meant
to let you
know that
I would do
that
BEFORE
we issue
the
next
set of method
approvals.
If they
are
the same
or only
have
insignificant
differences,
then
we will include
the
ASTM
method
as
an approved
method.
Legally,
it won’t
be
an
approved
drinking
water
method
until
we
publish
a notice
in
the Federal
Register.
It’s my opinion
that
if the Waters
methods
aren’t
easily available
from
Waters, then
you
can
easily
justify not
including
them in your
state
regulations.
Our
ATP coordinator
wasn’t
able
to
find
a
copy
of the
nitrate/nitrite
method
in his
files.
However,
he
is still checking
on
it.
I
have
the
GA Tech method.
I
can email
it to
you
on Monday.
If you
need it before
then,
you
can
go
to the e-docket
for
the 2007
Methods
Update
Rule.
I know the
method
is in the docket,
because
I
put
it there and
it is available
for
download
through
the docket
site.
I
wilIsee
if I can
find
out how
we
should
be
referencing
the GA Tech
method.
I thought
our
information
was correct
when
we
went final
on the rule.
Hope this
helps.
Pat
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
wrote:
To: Pat
Fair/CI/USEPA/US@EPA
From:
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
Date:
07/10/2008
06:O7PM
Subject:
Re:
Waters Methods
Thank
you.
That nails
it down.
I
will cite
it
as
an ASTM
method.
I
have
another
method
problem.
I
have
been
trying
to
obtain
a
copy
of
that
Ra-226/Ra-228
method
by
gamma-ray
spectometry
developed
by
Georgia
Insitute
of Technology.
The
“Environmental
Resources
Center”
has
been
disbanned
or
something,
so
that the
number
at
40
C.F.R.
141.74
is
no
longer
valid.
It
may have
become
the
Environmental Radiation
Center
or
something.
I
have placed
several
calls
and
e-mails
with
Bernd
Kahn
and
the
Center
in
an
attmpt
to
locate
the
method,
but no
luck
so
far.
Can you
help on
this
one
too?
file://C
:\Documents
and Settings\McCambM\Local
Settings\Temp\GW}
00002.HTM
11/26/2008
Page
2 of
5
Michael
J.
McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
>>> <Fair.Pat@epamail.epa.gov>
7/10/2008
7:43
M >>>
Mike,
Here’s
Waters
6508 method that
was
approved
for drinking
water.
Aren’ t
you limited
to methods
that are listed
as
approved
for drinking
water?
Based
on the info
at the
top of the method,
I’m guessing
this
may
now be
an ASTM method.
It was
evaluated under
the ATP
program, so
EPA was
given the
method
prior to the ASTM
process.
I don’t know
this
for
sure
and it
will
be
something I investigate
as
we
begin
putting together
our
next Expedited
Methods Approval
FR
action.
(If
it is the same
method,
we’ll
probably list
it in Appendix
A.)
(See
attached
file: Waters Method
D
6508, Rev
2EPA-HQ-OW-2003-0070-0063
.pdf)
As for the
other method,
it was approved
prior
to the 2007
methods
rule.
I don’t have a
copy of it, because
I wasn’t
involved
in the earlier
methods
rules. However,
I
have
asked our ATP
coordinator
to see if
it
is in the ATP
file. When I
hear back from
him, I’ll
let you know.
Hope this helps,
Pat
“Mike
McCambridge”
<mccambridge@±pc
To
b.
state . ii . us>
Pat Fair/CI/USEPA/US@EPA
cc
file://C:\Documents
and Settings\McCambM\Local
Settings\Temp\GW}00002.HTM
11/26/2008
Page3of5
07/08/2008
03:29
PM
Subj ect
Re:
Waters Methods
Whatever
you
can
do for
me
when you
get
back.
I
have
continued
to
look
into
this
today.
I am
convinced
that Method
6500
added to
SW-846
in
Update
IV in
the end
of 2007
is
the
method
you
have called
T1
Method
D6508”
from Waters.
See 73
Fed. Reg.
486
(Jan.
3,
2008)
http://www.epa.gov/SW846/pdfs/6500.pdf.
If
this
is true,
I will
likely
cite
the SW-846
version
of
the
method,
since
it
is
much
easier
to
obtain
than
the
method
from
Waters.
As described,
Waters
initially
acted
like
I
spoke
a
foreign
language
when
I asked
for
T
’Method
D6508.’
T
As
for
Method
B-lOll,
it
seems
to
distill
down
to me
needing
the title
to
the
document
in
which
the method
appears.
Talk to
you when
you
return.
Michael
J.
McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
>>>
<Fair.Pat@epamail.epa.gov>
7/8/2008
2:18 PM
>>>
Mike,
I’m
working
off
site
today,
so I
don’t
have
access
to
the
references
I
file
:1/C
:\Documents
and Settings\McCambM\Local
Settings\Temp\GW}
00002.HTM
11/26/2008
Page 4
of
5
need
to answer your
questions.
I should
have
copies
of the methods
that
were
added
to
40 CFR
141
as part
of the 2007
methods
update rule.
If
these
Waters
methods
are prior
to that,
I might not
be able
to help
you.
Unfortunately,
I
don’t
know who
might have
them
other
than Waters.
I’ll
see what
I can find
tomorrow
and
get
back to you.
Pat
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
wrote:
To: Pat Fair/CI/USEPA/US@EPA
From:
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
Date:
07/08/2008
01:57PM
Subject: Waters
Methods
I have tried
to obtain
copies of
the two Waters
methods
referenced
in 40
C.F.R.
141.23(k) (1)
for fluoride
and nitrite/nitrate
using
the
contact
information
included in
the
rule.
At first,
the Waters rep
could
not
locate
anything based
on the
EPA descriptions
included
in the
rule.
This
morning I
received
two documents
that
purport to
be the
methods.
The documents
raise questions
that
you
might answer
for me.
The
copy
of
Method B-lOll
sent
me
by Waters
is nearly identical
to
one
that I found
on the USEPA
website.
The only
difference
between
the
two
is that the
method
from the
USEPA wèbsite
is
headed
‘
T
Waters.”
The
document
it appears to
include
pages 13 through
17 from some
reference.
It is undated,
which
means
that
I cannot
use
it for
an
incorporation
by
reference.
Do you have
a dated copy
of Method
B-lOll or
a fuller
copy -
of
the posted
reference
that would
include
the date?
It
appears
that
the method
is just
one
cited out of
a fuller
reference, and
I should
cite to that
fuller
reference
by its own
title. I will
also
approach
Waters with
this
request.
Your
rule cites
“Waters Method
D6508,
Rev. 2,”
entitled
“Test
Method
file :1/C :\Documents
and
Settings\McCambM\Local
Settings\Temp\GW}
00002.HTM
11/26/2008
Page 5
of
5
f or
Determination
of
Dissolved
Inorganic
Anions
in Aqueous
Matrices
Using
Capillary
Ion
Electrophoresis
and
Chromate
Electrolyte.”
Waters
sent
me
a document
marked
TMethod
6500,”
“revision
0,”
and
dated
February
2007,
and
entitled
“Dissolved
Inorganic
Anions
In Aqueous
Matrices
By
Capillary
Ion
Electrophoresis.”
That
document
appears
to be
Method
6500
from
SW-846.
Is “Waters
Method
D6508,
Rev.
2” the
same as
Method
6500,
rev.
0 from
SW-846?
If
so, why
did
USEPA
cite
this as
“D6508”?
If
not,
can
you
forward
me a copy
of Method
D6508
or
give me
enough
information
to identify
the
method
to Waters,
that
I might
obtain
a copy
of
the
right
method?
Michael
J.
McCambridge
Attorney
Illinois
Pollution
Control
Board
312
-814-6924
fi1e://C:’Documents and Settings\McCambM\Local
Settings\Temp\GW}00002.HTM
11/26/2008
From:
Mike
McCambridge
To:
Fair.PatepamaiI.epa.gov
Date:
7/11/2008
3:45:30
PM
Subject:
Re: Waters
Methods
It does, as the stream
moves ever onward.
Wasn’t
it Aristotle who
said that
you cannot
step into the same stream
twice?
Michael
J. McCambridge
Attorney
Illinois Pollution Control
Board
312-814-6924
>>>
<Fair.Patepamail.ea.ov>
7/10/2008
9:31 PM
>>>
Mike,
I haven’t
done a one-to-one
check of the ASTM
method against the Waters
method,
so
I
can’t
say for sure that they are
the
same.
My comment
was
meant to let
you
know that
I would do that BEFORE
we
issue the
next set of method approvals.
If they
are the
same or only
have insignificant
differences, then we
will include the ASTM method
as an
approved method.
Legally, it won’t
be an
approved
drinking
water
method until we
publish a notice in the Federal
Register.
It’s my opinion
that
if the Waters
methods
aren’t
easily available from Waters,
then
you can easily
justify not including them
in your
state
regulations. Our ATP
coordinator
wasn’t
able to find
a copy of
the
nitrate/nitrite method
in his files. However,
he is still
checking on it.
I have the GA Tech method.
I can email
it to
you on Monday.
If you need it before then,
you can go to the
e-docket for the
2007
Methods
Update Rule. I know the method
is in the docket,
because I
put it there and it is available
for download through
the docket
site.
I will
see if I can find out
how we should
be
referencing
the GA Tech method.
I thought
our information was correct when
we
went
final on the rule.
Hope this
helps.
Pat
“Mike McCambridge”
<mccambridgeäipcb.state.il.us>
wrote:
To: Pat
Fair/Cl/USEPAIUS@EPA
From: “Mike McCambridge”
<mccambridoe(ircb.state.iI.us>
Date: 07/10/2008 06:O7PM
Subject:
Re:
Waters Methods
Thank
you.
That nails it down.
I
will cite it as an
ASTM method.
I have
another method problem. I have
been trying to
obtain
a copy
of that
Ra-226/Ra-228
method
by
gamma-ray spectometry
developed by Georgia
Insitute of Technology.
The
“Environmental
Resources Center”
has been disbanned or something,
so
that
the number
at 40 C.F.R. 141.74 is no longer
valid. It may
have become the Environmental
Radiation Center
or something.
I have
placed several calls and
e-mails with Bernd Kahn and
the
Center in an
attmpt to locate the method,
but no luck so far.
Can you help on this one
too?
Michael
J. McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
>>> <Fair.Pateamail.epa.ov>
7/10/2008 7:43
AM >>>
Mike,
Here’s Waters 6508
method
that
was approved
for
drinking water.
Aren’t
you
limited to
methods that are listed
as approved for drinking
water?
Based on the info at the
top
of
the method, I’m
guessing this may now
be
an
ASTM
method.
It was evaluated
under
the
ATP program, so
EPA was
given
the method prior to
the
ASTM process. I
don’t know this for sure
and it will be
something
I investigate
as
we
begin putting together our
next Expedited
Methods
Approval
FR
action. (If it
is
the
same method,
we’ll
probably list it in Appendix A.)
(See attached file: Waters Method
D 6508, Rev
2_EPA-HQ-OW-2003-0070-0063.pdf)
As for the other method,
it was approved prior
to the 2007 methods rule.
I don’t have
a
copy of it,
because I wasn’t involved
in
the earlier
methods
rules. However, I have
asked our ATP coordinator to
see if it
is in the
ATP file. When I hear back from
him, I’ll let you know.
Hope this helps,
Pat
“Mike
McCambridge”
<mccambridgeipc
To
b.state.il.us>
Pat
Fair/Cl/USEPAIUS@EPA
cc
07/08/2008
03:29
PM
Subject
Re: Waters
Methods
Whatever you can do for me when
you
get
back. I have continued to look
into this today. I am convinced that Method
6500 added
to
SW-846 in
Update IV in the end of 2007 is the
method you have called “Method
D6508”
from Waters. See 73 Fed. Reg. 486
(Jan.
3, 2008);
http://www.epa.qov/SW-846/pdfs/6500.pdf.
If this is true,
I will likely
cite the SW-846 version of the method,
since it is much easier to obtain
than the method from Waters. As described, Waters initially
acted like
I spoke
a
foreign
language when
I asked for “Method D6508.”
As
for
Method B-i 011, it seems to distill down to me needing the
title to the
document in which the method
appears.
Talk to
you
when
you
return.
Michael
J.
McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Patepamail.epa.qov>
7/8/2008 2:18 PM >>>
Mike,
I’m
working off site today, so I don’t have
access to
the
references I
need to answer your
questions.
I should have copies of the
methods
that
were
added
to 40 CFR 141
as
part of the
2007 methods update
rule. If
these Waters methods are prior to that, I might not
be able to help
you.
Unfortunately, I don’t know
who
might have them other than
Waters.
I’ll see
what I can find tomorrow and get back to
you.
Pat
“Mike McCambridge”
<mccambridcietipcb.state.iI.us>
wrote:
To: Pat
Fair/Cl/USEPA/US@EPA
From: “Mike McCambridge”
<mccambridgeticb.state.il.us>
Date:
07/08/2008 01:57PM
Subject: Waters Methods
have
tried
to obtain copies of the
two Waters methods
referenced
in 40
C.F.R.
141 .23(k)(1)
for
fluoride and
nitrite/nitrate
using the contact
information included
in
the
rule. At
first, the
Waters
rep could not
locate anything based on the
EPA descriptions
included in the rule.
This
morning
I received two
documents
that
purport
to be the methods.
The documents
raise questions
that
you
might answer
for
me.
The copy of Method B-lOll
sent me by Waters
is nearly identical
to one
that
I found
on the USEPA website.
The only difference
between the two
is that
the method
from
the USEPA website
is headed “Waters.”
The
document it appears
to include
pages
13 through
17
from some reference.
It is
undated, which means that
I
cannot use it
for
an
incorporation
by
reference. Do
you
have
a
dated
copy of
Method
B-I 011 or
a
fuller
copy
of the
posted reference that would
include the date?
It appears that
the method is just
one cited out of a fuller
reference, and I should
cite
to that fuller
reference by its own title.
I will also
approach
Waters
with this request.
Your rule cites
“Waters Method
D6508, Rev. 2,”
entitled “Test Method for
Determination
of Dissolved
Inorganic Anions
in Aqueous Matrices
Using
Capillary Ion Electrophoresis
and Chromate
Electrolyte.”
Waters sent me
a document marked “Method
6500,”
“revision 0,”
and dated February 2007,”
and entitled “Dissolved
Inorganic
Anions
In Aqueous
Matrices
By
Capillary
Ion Electrophoresis.” That
document appears
to be Method
6500
from SW-846. Is “Waters Method
D6508, Rev.
2” the same as Method
6500,
rev.
0 from SW-846?
If so, why did
USEPA cite this as “D6508”?
If not,
can
you
forward
me a copy
of Method D6508
or give me enough information
to identify the method
to Waters, that
I might obtain
a
copy of
the
right
method?
Michael
J. McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
From:
Mike McCambridge
To:
Fair.PatepamaiI.epa.gov
Date:
8/6/2008
2:08:48
PM
Subject:
Georgia
Radium Method
I am
having
trouble
locating
the new
method for
Ra-226
and
Ra-228
from the
Georgia
Institute
of Technology
that
USEPA
added on
March 12, 2007.
The
Environmental
Resource
Center may
no longer
exist. I need
a copy
of the method
if it is
to
appear
in the Illinois
regulations.
I
must also
assure that the
availability
information
is presented
correctly.
Can
you provide
a copy of
the method?
Can
you provide where
the public
may
obtain
the
method?
Tomorrow
the Board
will propose
the
amendments
that will
include the
March
12, 2007 methods
revisions
and the June
3,
2008
equivalent
methods approvals.
Issues
will remain
regarding
Waters
Method
6508,
rev. 2,
which
is
the
same
as ASTM
D6508-00(2005)e2,
since I
can get it
from ASTM but
not from
Waters
Corp., and
the Georgia
Radium
Method
that I now
seek.
Michael
J.
McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
Page 1
of
1
Mike
McCambridge
- Re:
Georgia
Radium
Method
From:
<Fair.Patepamai1.epa.gov>
To:
“Mike
McCambridge”
<mccambridgeipcb.state.il.us>
Date:
8/6/2008
10:04PM
Subject:
Re:
Georgia
Radium
Method
Mike,
I’m
on
travel
this week,
so
I don’t
have
access
to
the
GA Radium
method.
I’ll
send
it
to
you early
next
week.
I
seem
to remember
you
saying
that
the
contact
information
that was
given
to
us
for
the
March
2007
methods
rule
is no
longer
applicable.
I’ll have
to see
if
I can find
the
correct
contact
information
for
you.
I know
I can
give
you
the
method,
but I’m
sure
you still
need
a
source
to
publish
in
your
regulations.
Pat
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
wrote:
To:
Pat
Fair/CI/USEPA/US©EPA
From:
“Mike
McCambridge”
<mccambridge@ipcb.state.il.us>
Date:
08/06/2008
12:08PM
Subject:
Georgia
Radium
Method
I
am
having
trouble
locating
the
new
method
for
Ra-226
and
Ra-228
from
the
Georgia
Institute
of
Technology
that
USEPA
added
on
March
12, 2007.
The Environmental
Resource
Center
may
no longer
exist.
I need
a copy
of
the
method
if
it
is
to
appear
in the
Illinois
regulations.
I must
also
assure
that
the availability
information
is
presented
correctly.
Can
you
provide
a
copy
of
the
method?
Can
you
provide
where
the public
may obtain
the
method?
Tomorrow
the
Board
will
propose
the
amendments
that
will include
the
March
12,
2007
methods
revisions
and
the
June
3,
2008
equivalent
methods
approvals.
Issues
will
remain
regarding
Waters
Method
6508,
rev.
2,
which
is the
same
as
ASTM
D6508-
00(2005)e2,
since
I
can get
it
from
ASTM
but
not
from
Waters
Corp.,
and the
Georgia
Radium
Method
that
I
now
seek.
Michael
J.
McCambridge
Attorney
Illinois
Pollution
Control
Board
312-814-6924
file://C:\Documents
and
Settings\McCambM\Local
Settings\Temp\GW}00002.HTM
11/26/2008
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
9/11/2008
4:29:42
PM
Subject:
Radium
Method
I did
not receive
the
e-mail.
My IT
people tell me
that they
have
no way to recover
items
caught
in their
filters; they
do not
maintain
a “spam
folder,” as
appears on both
of
my
personal
e-mail accounts.
Try
one more time,
and use
this
address.
Please
CC
my
personal
e-mail
account:
m.mccambridgeiatt.net.
Also
use
your
EPA
address,
and
give
the
IT people
here a couple
of
days
to
make the necessary
system
adjustments.
I
am
sending
a copy
of
this e-mail
to
our IT
people, and
I will
ask them
to
include
your
domain
as aIIowed.”
It
would
amaze me
if USEPA
is blocked
as
a domain, but
anything
is
possible.
We live
in such
a nightmare
world
of
spam
and malicious
e-mail. The best
efforts
of the most
conscientous
IT
protocol is
bound
to
“gang aft agley,”
as
Rabbie
Burns would have
it.
If you
have trouble,
call me.
Michael
J. McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
From:
Mike
McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
9/15/2008
12:02:52
PM
Subject:
Geargia
Tech
Radium
Method
I did receive
the
method, but
only
on
my personal
e-mail account,
not on the
State system.
We must bear
this in mind
for
the future.
If
you
need to contact
me, use
my personal e-mail
(m.mccambrideatt.net)
or
call
me.
Thanks
for your efforts.
I have
only one
minor favor
remianing
to
ask:
could
you
let
me know what
you
learn
with
regard to
availability. I
will
redouble
my efforts to
get
this
information
myself,
and I will
let
you
know
if
I
learn
enything,
but
I will need that
information
to
complete
the incorporation
by reference.
Perhaps,
“U.S.
EPA”
will get
a response
before
“Pollution Control
Board.”
So far,
I
have received
no responses
to calls or e-mails.
Michael
J.
McCambridge
Attorney
Illinois
Pollution Control
Board
312-814-6924
From:
Mike McCambridge
To:
Fair.Pat©epamail.epa.gov;
TURLEY, Dawn
Date:
9/17/2008
9:06:17
AM
Subject:
Re: Test for
IPCB E-mail delivery
It
worked, thank
goodness.
Thank
you both
(Pat Fair and Dawn
Turley) for all your help.
Michael
J. McCambridge
Attorney
Illinois Pollution
Control
Board
312-814-6924
>>> <Fair.Pat@epamail.epa.gov>
9/17/2008
8:06 AM >>>
I
added Mike’s name to
my email list.
I believe
the earlier messages
that I sent to him
were either replies
to his messages
or I
copied/pasted
his address from
an earlier
email.
Hope you figure out
the problem. You
can see if he receives
this message,
since
he’s copied
on it.
“Dawn
TURLEY”
<turleydicb.st
ate.il.us>
To
Pat
Fair/Cl/USEPAJUS@EPA
09/16/2008 10:17
cc
AM
Subject
Re: Test for IPCB
E-mail delivery
Thank you
for
responding
so
quickly.
I am trying to determine
why Mike
is
not getting your
e-mails. Would you please
remove Mike McCambridges
e-mail from
your
address book and recreate
his address to
ensure that
it’s correct?
His address is
mccambmipcb.state.il.us
Thank
you,
Dawn Turley
IPCB
Network Support
Phone: 217-782-2415
Fax: 217-524-8508
On 9/15/2008
at 7:25 PM, <Fair.Pat(5epamail.epa.qov>
wrote:
Dawn,
I received your email.
Pat Fair
“Dawn TURLEY”
<turleyd(ircb.state.il.us>
wrote:
To:
Pat
Fair/Cl/USEPAIUS@EPA
From: “Dawn
TURLEY”
<turleydipcb.state.iI.us>
Date:
09/15/2008 02:27PM
Subject:
Test
for IPCB E-mail
delivery
I
am the e-mail
administrator
for the IPCB.
Mike McCambridge
is
having
problems receiving
your e-mails.
In order
to track
and
resolve
the
problem,
would
you
please
reply to
this e-mail
and then
try to
send me
a
new
e-mail?
Thank
you,
Dawn
Turley
IPCB
Network
Support
Phone: 217-782-2415
Fax: 217-524-8508
From:
<Fair.Pat©epamail
.epa.gov>
To:
MCCAMBM©ipcb.state.il.us
Date:
10/15/2008
10:30:16AM
Subject:
Source
of Radium
226-228
method
Mike,
I finally
tracked
down
the contact
information
for
obtaining
the GA Tech
method
for radium
226 &
228 in drinking
water.
Here it is:
Robert
Rosson
Georgia Tech
Research
Institute
925
Dalney
Road
Atlanta, GA
30332
(404)407-6339
robert.rossongtri.gatech
.edu
I
can’t
easily
update
the
CFR
to reflect
this
changed
contact
information.
I am going
to
try to
update
the information
on
EPA’s
drinking
water web
page,
so it will
be accurate.
I hope this
information
helps
and
is
not too late.
Pat Fair
From:
Mike McCambridge
To:
Fair.Pat©epamail.epa.gov
Date:
10/15/2008 10:32:50AM
Subject:
Re:
Source of
Radium 226-228
method
Thank
you
very much. I now have
all I need
to
include
the method
in the
pending update, so that entities
in
Illinois may
opt
to
use the method.
Michael
J. McCambridge
Attorney
Illinois Pollution Control Board
312-814-6924
>>> <Fair.Patepamail.epa.gov>
10/15/2008
10:29
AM
>>>
Mike,
I finally tracked down the
contact information
for obtaining the GA Tech
method for
radium
226
& 228 in drinking
water. Here it is:
Robert Rosson
Georgia
Tech Research Institute
925 Dalney Road
Atlanta,
GA
30332
(404)407-6339
robert.rossonctri.qatech.edu
I can’t
easily update the CFR to reflect
this changed contact
information. I
am
going to try
to
update the information
on EPA’s
drinking water
web page, so it will
be
accurate.
I
hope this information
helps and is
not too late.
Pat Fair
United States
EnvironmGntal Protection
Agency
Office of Research
and
Development
Washington
DC 20460
EPAI600JR-94/1 73
October 1994
•6EPA
/
Technical
Notes
C)fl
Drinking
Water
Methods
7Z
-A16
-
/4/
AC
9
S
EPAJ600)R-941173
October
1994
TECHNICAL
NOTES
on.
DRINKING
WATER
METEIODS
U.
S. Environmental
Protection
Agency
Office
of Water
Office
of Ground
Water
and
Drinking
Water
Office
of
Research
and
Development
Environmental
Monitoring
Systems Laboratory
Cincinnati,
01145268
Printed
On
Recycled
Paper
DISCLAIMER
This
manual has
been
reviewed
by
the
Technical
Support
Division,
Office
of
Water
and the
Environmental
Monitoring
Systems
Laboratory
—
Cincinnati,
U.S.
Environmental
Protection
Agency,
and
approved
for
publication.
Mention
of
trade
names or
commercial
products
does
not
constitute
endorsement
or
recommendation
for
use.
ii
0
FOREWORD
‘Compliance
with
National
Primary
and
Secondary
Drinking
Water
Regulations
requires
that
analyses
of
samples.
be
conducted
by
a
certifiedlaboratory.
A
certification
condition
is
that
an
approved
method
be
used.
The
Office
of
Water’s
(OW)
Technical
Support
Division
(TSD)
prepares
the
analytical
methods
parts
of
drinking
water
regulations.
The
Office
of
Research
and
Development’s
(ORD) Environmental
Monitoring
Systems
Laboratory
at
Cincinnati,
Ohio
(EMSL—
Cincinnati)
conducts
research
to
develop
and
evaluate
analytical
methods
for
the
determination,
of
contaminants
in
many
media
including
drinking
water.
EMSL—Cincinnati
also
regularly
publishes
methods
for
use
in
drinking
water
compliance
monitoring.
This joint
OW/ORD
publication,
Technical
Notes
on
Dr:inking
Water
Methods,
was
prepared
to
add
modifications,
clarifications,
options
or
improvements
to
methods
that have been
previously
approved
and
published.
To
allow
the
public
to
use
these
changes
without
waiting
for
incorporation
in
the
next
revision
of
a
method,
EPA
has elected
to
describe
the
changes
in
this
document.
The
Office of
Water will approve
these
changes
in
a
1994
rulemaking
by
incorporating
Technical
Notes
on
Drinking
Water
Methods
into
the
drinking
water regulations.
Procedures
described
herein
supersede
or
complement
procedures
described
in
the
approved
methods.
When
a
method
is
revised,
relevant
procedures
from
this
document
will
be
included
in
the
revised
method.
We
are
pleased
to
provide
these
technical
notes
and
believe
they
will
be
of
considerable
value to
public
and
private
laboratory,
rgulatory
and
certification
personnel.
Alan
A.
Stevens,
Director
Thomas
Clark,
Director
Technical Support
Division
Environmental
Monitoring
Systems
Office
of
Water
Laboratory
—
Cincinnati
111
Table
of
Contents
TITLE
PAGE
Disclaimer
.11
Foreword
Acknowledgments
Introduction
vii
I.
Approved Drinking Water Methods for Compliance Monitoring
I
II.
Methods
To Be Withdrawn
on July 1, 1996
14
III.
Recommended
Methods for
Secondary Drinking Water Contaminants
20
IV.
Mandatory Method Modifications
22
Standard Method 4500—Cl—E
(Chlorine Residuals)
23
Standard Method 4500—Cl—G
(Chlorine Residuals)
24
Protocol
for Continuous Chlorine
Residual Monitoring
25
Spectrophotometric Determinations of Cyanide
26
Turbidimeter Calibration
28
Sample Digestion for Determination of
Metal Contaminants
29
Standard Method 3114B (Arsenic and Selenium)
30
Standard Method 3113B
and ASTM D3859—93B
(Selenium)
31
Standard Method 3113B
(Chromium)
32
EPA
Methods502.2
and 524.2, Sorbent
Traps
33
EPA
Methods 502.2, 524.2
and 551, Sample Acidification
34
EPA
Method 506, Errata
in Summary
35
EPA
Method 508,
DCPA and
Hexachiorocyclopentadiene
36
EPA
Methods 515.1 and 515.2, Use
of
TMSD
37
iv
0
Table of Contents
TITLE
PAGE
IV. Mandatory
Method Modifications (Continued)
EPA
Method 524.2,
Quality
Assurance, VOC Data
40
EPA Method
531.1 and SM 6610,
Storage of Samples
44
EPA
Method
551,
Pentane
45
EPA
Method549.1, Sample
Containers
.46
Alternative Liquid—Solid Extraction Cartridges and
Disks......47
V.
Recommended Method Modifications
48
EPA
Method 100.1, Asbestos
Guidance
49
EPA Method 502.2, Use of thePID
52
EPA Methods 502.2, 524.2
and
551, Sample
Dechlorination
EPA
Method 504.1, hromatographic Interferences
.54
EPA
Methods
505, 507, 508,
InterChange of Detectors
56
EPA
Methods 507, 508 and 515.1,
Mercuric Chloride...
..
57
EPA Method
1613, Dioxin
Guidance
58
VI.
EPA
Contacts
and Method References
59
V
(U
ACKNOWLEDGMENTS
We
appreciate
the many
constructive
comments
and
informative
questions.
from
our
customers,
the
analytical
and
certification
laboratory
community.
Their
information provided
the
basis
for
the
options,
clarifications
and
method
modifications
that
are
approved
and
described
in
these
technical
notes.
Many
people
in
the
Office
of Research
and
Development’s
Environmental
Monitoring Systems
Laboratory
— Cincinnati
(EMSL—Cincinnati)
and
in
the
Office
of
Ground
Water
and
Drinking
Water’s
Technical
Support
Division
(TSD)
in
Cincinnati
contributed
to these
notes.
The
EPA
scientists
in
these
groups
used
information from
their
many
contacts
with
the
public,
and
their
years
of
experience
with
drinking
water
analysis
to
produce
this
publication.
Technical Notes
was
developed
and edited
by
Richard
Reding
of
TSO
who.
wishes
to
especially
acknowledge
the
contributions
of
Thomas
Behymer,
James
Eichelberger,
Theodore
Martin,
Jean
Munch,
James
O’Dell,
John
Pfaff,
Jody
Shoemaker
and
Nancy
lilmer
from
EMSL—Cincinnati,
Patricia
Snyder
Fair,
Marianne
Feige, Edward
Glick,
David
Munch
and
Kent
Sorrell
from
TSD,
and
Patrick
Clark
from
the
Risk
Reduction
Engineering
Laboratory
in
Cincinnati.
Carol
Madding,
TSD,
contributed
technical
notes
and
helped
with
the
editorial
design.
In
addition,
the
names
of
the
developers
of
the
methods
and
instrumentation
that
are
the
subject
of
this
publication
can
be
found
in
the
acknowledgment
and
reference sections
of
the
EPA
method
or
EPA
methods
manual.
The
administrative
personnel
of
EMSL—Cincinnati,
in
particular
Diane
Schirmann, Patricia
I-Iurr,
and
Helen
Brock,
provided
outstanding
support
to
this
effort.
The
editor
also
thanks
the
administrators
and
managers
of
the
Environmental Protection
Agency
who
supported
the
development
and
preparation
of
this
document.
Special
appreciation
is
due
to
Herbert
J. Brass,
Chief
of
the
Drinking Water
Quality
Assessment
Branch,
TSD,
and
William
L. Budde,
Director
of
the
Chemistry
Research
Division,
EMSL—Cincinnati,
for
their
cooperation and
support during
this
project.
vi
0
INTRODUCTION
Richard
Reding
This document,
Technical
Notes
on
DrinkingWater
Methods,
describes
method
modifications
that
were
developed
after
an
approved
method
had
been
published.
Most
of
the
modifications
were
formerly
footnoted
in
the
drinking
water
regulations,
or
were
described
in
a proposed
rule
(58
fj3,
65622,
December
15,
1993). Because
this
document
is
incorporated
by
reference
in
drinking
water
regulations,
it
is
a
mandatory
part
of
the
analytical
procedures
required
to
conduct
complian.àe
monitoring
and
to
obtain
laboratory
certification.
Laboratories
can
use.
this
publication
as
a
guide
to
analytical
methods
approved
under
the
Safe
Drinking
Water
Act
(SDWA),.to
obtain
information
on
the
latest
approved
modifications.to
these
methods,
and
to
contact
EPA
with
questions
about
drinking
water
methods.
Since
EPA
method
manuals
are
printed
in
a
looseleaf
format,
the
format
of
Technical
Notes
allows
readers
to
insert..
pages containing
a
method
change
in
the
manual
containing
the
áffected•EPA
analytical
method.
Methods
approved,
for
monitorin.g
unde.r
National
Primary
Drinking
Water.
Regulations
are
in
Section
I
of
this
document...
Methods
for
which
‘approval
will
be
withdrawn
in
1996
are
in
Section
II,
and
methods
for
monitoring
under
.
National
Secondary
Drinking
Water
Regulations
are
contained.in
Section
III.
Mandatory
method
modifications
are
described
in
Section
IV.
The
modifications
include
a
protocol
for
monitoring
chlorine
residuals
continuously
as
required
under the
Surface
Water
Treatment
Rule,
requirements
for
mandatory
nranual
distillation
of
samples
collected
for
determination
of
cyanide,
and
use
of
another derivatizing
reagent
with
EPA
Methods
515.1
and
515.?.
Technical
notes
on
optional
procedures
and
recommended
modifications
to
compliance
methods
are
described
in
Section
V.
. These notes
include
guidance
on
how
to
make
analyses
of
asbestos-
and
dioxin
more
cost—effective,,
and
when
to
omit
use
of
mercuric
chloride
in
some
EPA
pesticide
methods.,
The
remainder
of
this
introduction
provides
guidance
on
methods
selection
and
on
the
laboratory
certification
aspects
of
approved
methods.
SELECTION
OF
METHODS FOR
OTHER
CHEMICALS
EPA
believes
that
some
water
systems
wish
to
measure
‘bhemicals
that
are
not
included
in
drinking
water
regulations,
and
need
advice
on
what
method
to
use.
The
December
1993
Proposal
noted
that
while
EPA
only approves
methods
for
contaminants
regulated
under
the
SDWA,
the
Agency
encourages
laboratories
to’
use
these methods
for
voluntary
monitoring
of
other
contaminants,
“if
the
method
description
specifically
includes
these.
contaminants.”
This
recommendation
does
not
preclude
use
of
other
methods,
including
test
kits,
for
voluntary
monitoring.
Analysts
always should
carefully
evaluate
the
performance
of
any
method
when
using
it
for
samples
other
than
compliance
monitoring
samples,
or
for
contaminants
not
regulated
under
the
SDWA.
vii
LABORATORY
CERTIFICATION
When
using an approved
method
to obtain certification
or tà
conduct
compliance
monitoring,
EPA strongly encourages
users
of methods that
are
published
in an EPA manual
to follow
instructions contained
in the
introductions to
these manuals,
unless
the
instructions
conflict
with
statements
in
this document,
or
in
the
drinking water
regulations.
Although
must”
can be
argued to
be
a
stronger word
than “should”
in
requiring
adherence
to
method
procedures,
some
approved
methods
use
these
terms
interchangeably.
Analytical methods
for drinking
water
are written to
be
prescriptive
enough
to provide
uniformity of data
quality,
and flexible
enough
to allow analysts
to
exercise judgment,
skill
and
initiative
to
improve
the
overall quality
and efficiency
of compliance
monitoring.
The Agency
does not
believe
that
semantical
differences between
“must” or
“should”
limits the
authority
of certification
officials to
enforce provisions
of
the
methods.
viii
0
SECTION
I.
APPROVED
DRINKING
WATER
METHODS
FOR
COMPLIANCE
MONITORING
To
make
this
document
a
more
complete
source
of
current
methods
information,
the
approved
methods
which
are
specified
in
regulations
at
40
CFR
Part
141,
are
listed
in
this
section.
Methods
for
which
approval
will
be
withdrawn
in
1996
are
in
Section
II.
Recommended
methods
for
secondary
contaminant
monitoring,
which
are
specified
in
regulations
at
40
CFR
Part
143,
are
listed
in
Section
III.
1
METHODS FOR COLIFORM
SAMPLING
To
comply with the provisions of the Total Coliform Rule, public water
systems
must conduct analyses in
accordance
with
one of the analytical
methods
in the following table.
Total coliform methods, except
for
the Colisure Test,
are contained in
the
18th
edition of Standard Methods for the Examination
of
Water
and
Wastewater,
1992, American Public health Association, 1015
Fifteenth
Street
NW,
Washington,
D.C.
20005. Preparation of the
EC medium
and the
nutrient agar are
described
in
Standard Methods,
p.
9—52,
para. la, and
pp.
9—
47
to 9—48, respectively. A description of the Colisure Test maybe obtained
from the Millipore Corporation,
Technical
Services Department, 80 Ashby Road,
Bedford, MA
01730. The phone number is (800) 645—5476.
Organism
Methodology
Citation
Total
Coliforins
1
Total Coliform Fermentation
9221A,
B
Techni
que
2’
3’
4
Total Coliform Membrane
Filter
9222A,
B, C
Technique
Presence—Absence
(P—A)
Coliform
9221D
Test
4’5
ONPG—F4UG Test
6
9223
Colisure
Test
7
Footnotes
The time
from sample collection to initiation
of analysis may not exceed
30
hours.
2
Lactose
broth, as commercially available, may
be
used in lieu of
lauryl
tryptose broth, if the
system conducts
at least 25 parallel tests between
this
medium
and lauryl
tryptose broth using
the
water normally tested, and
this
comparison demonstrates
that the false—positive
rate
for total coliforms,
using
lactose
broth, is
less
than
10 percent.
If
inverted
tubes are used
to
detect gas production,
the media should cover
these tubes at
least
one—half
to two—thirds
after the sample is added.
‘
No requirement
exists
to run
the completed
phase On 10 percent of all
•total
col
iform—positive confirmed
tubes.
Six—times
formulation strength
may
be used if the
medium is filter—
sterilized
rather than autoclaved.
.
6
The
ONPG—MUG Test is also known as the Autoanalysis
Colilert System.
The
Colisure
Test
must be incubated for 28 hours before
examining the
results.
If examination at
28
hours is not
convenient, then results
may be
examined
at any.
time
between 28 hours and 48 hours.
2
.
.
.
METHODS
FOR
INORGANIC
CHEMICALS
AND
OTHER
PARAMETERS
Analysis
for
the
following
contaminants
shall
be
conducted
in
accordance
with
the
methods
in
the
following
Table,
or
their
equivalent
as
determined
by
EPA.
The
monitoring
requirements
for
these
contaminants
are
specified
at
§
141.23,
141.41,
and
141.80
141.91.
Criteria
for
analyzing
arsenic,
barium,
beryllium,
cadmium,
calcium,
chromium,
copper,
lead,
nickel,
selenium
and
thallium
with
digestion
or
directly
without
digestion,
and
other
mandatory
procedures
are
contained
in
Section
IV
of
this
Technical
Notes
document.
Guidance
on
conducting
asbestos
analysis
is
described
in
Section
V
of
Technical
Notes.
Contaminant
Methodology
EPA
ASTM
Other
Antimony
ICP-Mass
Spectrometry
2OO.8
Hydride—Atomic
Absorption
D—3697—92
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
3113B
Arsenic
Inductively
Coupled
Plasma
2OO.7
3120B
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
D—2972—93C
31138
Hydride
Atomic
Absorption
D—2972—93B
3114B
Asbestos
Transmission
Electron
Microscopy
ioO.i
Transmission
Electron
Microscopy
lOO.2
Barium
Inductively
Coupled
Plasma
2OO.7
3120B
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Direct
3111D
Atomic
Absorption;
Furnace
3113B
Beryllium
InductivelyCoupled
P1asma_
-
312OB
ICP-MassSpectrometry
2O0.8
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
D—3645—93B
31138
Contaminant
Methodology
EPA
Other
Cadmium
Inductively
Coupled
Plasma
2OO.7
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
3l13B
Chromium
Inductively
CoupledPlasma
2OO.7
3120B
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
3113B
Cyanide
Manual
Distillation
followed
by
4500-’CN—C
Spectrophotometric,
Amenable
D2036-9lB
4500—CN--G
Spectrophotometric
Manual
D2036—9lA
4500—CN—E
I_3300_856
Semi—automated
33547
Selective
Electrode
4500CN-F
Fluoride
Ion
Chromatography
3OO.O
D4327-9l
41105
Manual
Distill.;
Color.
SPADNS
4500F—B,D
Manual
Electrode
D1179—93B
4500F—C
Automated
Electrode
380-75WE
8
Automated
Alizarin
4500F—E
129-71W
8
Mercury
Manual,
Cold
Vapor
24S.l
03223—91
3ll2B
Automated,
Cold
Vapor
245.2
•
ICP—Mass
Spectrometry
200.8
Nickel
Inductively
Coupled
Plasma
2O0.7
31205
ICP—Mass
Spectrometry
20O.8
Atomic
Absorption;
Platform
2O0.9
Atomic
Absorption;
Direct
3lllB
Atomic
Absorption;
Furnace
3l13B
Nitrate
Ion
Chromatography
•
3OO.0
D4327—9l
41105
B-lOll’
0
Automated
Cadmium
Reduction
353.2
D3867—90A
4500—N0
3
—F
Ion
Selective
Electrode
4500—N0
3
—D
601”
Manual
Cadmium
Reduction
D3867—90B
4500—N0
3
—E
Q
..
0
Contaminant
Methodology
EPA
ASTM’
SM
2
Other
-
Nitrite
Ion
Chromatography
3OO.O
D4327—91.
4110B
B-lOll’
0
Automated
Cadmium
Reduction
353.2
03867—90A
4500—N0
3
—F
Manual
Cadmium
Reduction
D386790B
4500—N0
3
E
Spectrophotometric
4500—NO
2
—B
Selenium
Hydride—Atomic
Absorption
D3859—93A
31148
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Platform
2OO.9
Atomic
Absorption;
Furnace
.
03859—93B
3113B
Thallium
ICP—Mass
Spectrometry
2OO.8
Atomic
Absorption;
Platform
2OO.9
Lead
Atomic
absorption;
furnace
D3559—90D
3113B
ICP—Mass
spectrometry
2OO.8
Atomic
absorption;
platform
2OQ.9
Copper
Atomic
absorption;
furnace
D1688—90C
3113B
Atomic
absorption;
direct
aspiration
D1688—90A
3111B
ICP
2OO.7
3120B
ICP
—
Mass
spectrometry
2OO.8
Atomic.
absorption;
platform
2OO.9
pH
Electrometric
l5O.l
D1293—84
45OO—H—B
15O.2
-
Conductivity
Conductance
01125—91A
25108
Calcium
EDTA
titrimetric
D511—93A
3500—Ca—D
Atomic.
absorption;
direct
aspiration
D5l1—93B
3111B
Inductively—coupled
plasma
2OO.7
3120B
AIkällxuity
Titrirnetric
.
D1067—92B
2320B
Electrometric
titration
I1O3O85
Contaminant
Methodology
EPA
ASTM
1
SM
2
Other
Ortho—
Colorimetric,
automated,
ascorbic
acid
365.11
4500—P—F
phosphate
unfiltered,
Colorimetric,
ascorbic
acid,
single
D515—88A
4500—P—E
no
digestion
reagent
or
hydrolysis
Colorimetric,
phosphomolybdate;
I_1601_856
automated-segmented
flow;
I_2601_906
automated
discrete
I_2598_856
Ion
Chromatography
300.0
D4327—91
4110
Silica
Colorimetric,
molybdate
blue;
I_1700_856
automated—segmented
flow
L_2700_856
Colorimetric
D859—88
Molybdosilicate
4500—Si—D
Heteropoly
blue
4500—Si-E
Automated
method
for
molybdáte—reactive
silica
4500—Si—F
Inductively—coupled
plasma
200.7
3120B
Temperature
Thermometric
-
2550B
Sodium
Inductively—coupled
plasma
200.7,
Atomic
absorption;
direct
aspiration
3111B
e
Footnotes
Annual
Book
of
ASTM
Standards,
Vols.
11.01
and
11.02,
American
Society
for
Testing
and
Materials,
1916
Race
Street,
Philadelphia,
PA
19103.
18th
edition
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
1992,
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
“Methods
for
the
Determination
of
Metals
in
Environmental
Samples
—
Supplement
I”,
EPA—600/R—94/111,
May
1994.
Availalble
at
NTIS,
PB94—
184942.
Method
100.1,
“Analytical
Method
For
Determination
of
Asbestos
Fibers
in
Water,”
EPA—600/4—83—043,
September
1983
Available
at
NTIS,
PB83—
260471.
I
Method
100.2,
“Determination
Of
Asbestos
Structures
Over
10
j.Lm
in
Length
in
Drinking
Water,”
EPA/600/R—94/134,
June
1994.
Available
at
NTIS,
PB94—201902.
S
Available
from
BOktdOWZFiIë
Róftéctioñ
U.S.
Geological
Survey,
Federal
Center,
Box.
25425,
Denver,
CO
80225-0425.
“Methods
for
the
Determination
of
Inorganic
Substances
in
Environmental
Samples,”
EPA—600/R—93/100,
August
1993.
Available
at
NTIS,
PB94-
121811.
8
Industrial
Method
No.
129—71W,
“Fluoride
in
Water
and
Wastewater,”
December
1972,
and
Method
No.
380—75WE,
“Fluoride
in
Water
and
Wastewater,”
February
1976,
Technicon
Industrial
Systems,
Tarrytown,
NY
10591.
Methods
150.1,
150.2
and
245.2
are
available
from
USEPA,
EMSL—
Cincinnati,
OH
45268.
The
identical
methods
are
also
in
“Methods
for
Chemical
Analysis
of
Water
and
Wastes,”
EPA—600/4—79/020,
March
1983.
,
Method
B—lOll,
“Waters
Test
Method
for
Determination
of
Nitrite/Nitrate
in
Water
Using
Single
Column
Ion
Chromatography,”
Millipore
Corporation,
Waters
Chromatography
Division,
34
Maple
Street,
Milford,
MA
01757.
11
Technical
Bulletin
601
“Standard
Method
of
Test
for
Nitrate
in
Drinking
Water,”
July
1994,
PN
221890—001,
ATI
Orion,
529
Main
Street,
Boston,
MA
02129.
This
method
is
identical
to
Orion
WeWWG/5880,
which
is
approved
for
nitrate
analysis.
ATI
Orion
republished
the
method
in
1994,
and
renumbered
it
as
601,
because
the
1985
manual
“Orion
Guide
to
Water
and
Wastewater
Analysis,”
which
contained
WeWWG/5880,
is
no
longer
available.
7
METHODS FOR ORGANIC CHEMICALS
Analyses for regulated organic
contaminants under the
monitoring
requirements
specified
at
§141.24
and 141.30
shall be conducted using
the
following
EPA
methods
or their
equivalent as approved by
EPA. Other
mandatory
and optional procedures for
conducting
these methods
are described
in Sections
IV and V, respectively,
of
this
document.
Contaminant
Method
524.2
524.2,
524.2
524.2
524.2
524.2
524.2
524.2
524.2
524.2
524.2
524.2
524.2, 551
524.2, 551
524.2, 551
524.2
524.2
524.2
524.2
524.2
524.2
551
Benzene
Carbon
tetrachi oride
CM
orobenzene
1,
2—Di chi
orobenzene
1,4—Dichlorobenzene
1,2—Dichioroethane
ci s—Di chi
oroethyl
ene
trans—Di chi oroethyl ene
Dichi
oromethane
1,2—Dichloropropane
Ethyl benzene
Styren e
Tetrachi oroethyl
ene
1,1, 1—Tn
chi
oroethane
Tn
chi oroethyl
ene
Tol uene
1,2, 4—Tn chi
orobenzene
1, 1—Di
chi oroethyl
ene
1, 1,
2—Tn chi oroethane
Vinyl
chloride
Xylenes (total)
2,3,7,8—TCDD
(dioxin)
2,4—D
2,4,5—TP
(Silvex)
Alachior
Atrazine
Benzo(a)pyrene
Canbofuran
Chlordane
Dalapon
Di
(2—ethyl hexyl
)
adi pate
Di (2—ethyl
hexyl
)
phthal ate
Di brornochi
oropropane (DBCP)
Dinoseb
Diquat
Endothall
Endri
n
Ethylene
dibromide (EDB)
Gi
yphosate
Heptachlor
Heptachior
Epoxide
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
.502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
502.2,
1613
515.2, 555, 515.1
515.2, 555, 515.1
5051,
507, 525.2, 508.1
5051,
507,
525.2, 508.1
525.2, 550,
550.1
531.1, 6610
505,. 508,
525.2, 508.1
552.1,
515.1
506,
525.2
506, 525.2
504.1,
551
515.2,
555, 515.1
549.1
548.1
505,
508,
504.1
551
547,
6651
505, 508, 525.2,
508.1
505, 508, 525.2,
508.1
525.2, 508.1
8
.
Contaminant
Method
1-lexachi
orobènzene
Hexachi
orocyci
opentadi
ene
Lindane
Methoxych.lor
Oxamyl
PCBs
(as
decachlorobiphenyl)
2
(as
Aroclors)
Pentachi
orophenol
Picloram
Simazine
Toxaphene
Total
Trihalomethanes
508.1
508.1
508.1
508:.
1
505,
508,
525.2,
505,
525.2,
508,
505,
508,
525.2,
.505,
508,
525.2,
531.1,
6610
508A
505,
508
515.2,
525.2,
555,
515.1
515.2,
555,
515.1
5051,
507,
525.2,
508.1
505,
508,
525.2
502.2,
524.2,
551
1
A
nitrogen—phosphorous
detector
should
be
substituted
for
the
electron
capture
detector
in
Method
505
(or
another
approved
method
should
be
used)
to
determine
alachior,
atrazine
and
simazine,
if
lower
detection
limits
are
required.
2
PCBs
are
qualitatively
identified
as
Aroclors
and
measured
for
compliance
purposes
as
decachlorobiphenyl
using
Method
508A.
Methods
502.2,
505,
507,
508,
508A,
515.1
and 531.1
are
in
Methods
for
the
Determination
of
Organic
Compounds
in
Drinking
Water,
EPA—600/4—88—039,
Revised,
July
1991.
Methods
506,
547,
550,
550.1
and
551
are
the Determination
of
Organic
Compounds
in
Drinking
Water
—
December
1988,
in
Methods
for
Supplement
I,
EPA/600—4—90/020,
July
1990.
549.1,
552.1and
555
are in
Methods
for
the
in
Drinkina
Water
—
Sunolement
II
-
EPA/600/R—92/129,
August
1992.
Method
1613
is
titled,
“Tetra—Through
Octa—Chlorinated
Dioxins
and
Furans
by
Isotope
Dilution
HRGC/HRMS,!r
EPA
821—B—94—005,
October
1994.
These
documents
are
available
from
the
National
Technical
Information
Service,
(NTIS)
PB91—231480,
PB91—146027,
PB92—207703
and
PB95—104774,
U.S.
Department
of
Commerce,
5285
Port
Royal
Road,
Springfield,
Virginia
22161.
The
toll—free
number
is
800—553—6847.
EPA
Methods
504.1,
508.1
and
525.2
are
available
from
USEPA
EMSL—Cincinnati.,
Cincinnati,
OFF
45268.
The
phone
number
is
(513)—569—7586.
Method
6651
is contained
in the
18th
edition
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
1992,
and
Method6610
is
contained
in the
Supplement
to the
18th
edition
of Standard
Methods
for
the
Examination
of
Footnotes
Methods
515.2,
524.2,
548.1,
Determination
of
Oroanic
Comnounds
Water
and
Wastewater,
1994,
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
9
METHODS FOR
UNREGULATED
CONTAMINANTS
Regulations
specified
in
§141.40
require monitoring
for certain
contaminants
to
which
maximum
contaminant levels do not apply.
These,
chemicals are
called “unregulated” contaminants, and
presently include
sulfate,
34 volatile
organic chemicals (VOCs) and 13 synthetic organic
chemicals
(SOCs).
1.
Analysis
for
the
34 unregulated VOCs listedunder
paragraphs (e) and
(j)
of
§141.40 shall be conducted using
the following
recommended
methods,
or
their
equivalent as determined by EPA.
VOC
Contaminants
Method
Chloroform
502.2,
524.2,
551
Bromodichioromethane
502.2, 524.2, 551
Bromoform
502.2,
524.2, 551
Chiorodibromomethane
502.2, 524.2, 551
Bromobenzene
502.2, 524.2
Bromochioromethane
502.2, 524.2
Bromomethane
502.2,. 524.2
n—Butylbenzene
502.2, 524.2
sec—Butylbenzene
502.2, 524.2
tert—Butylbenzene
502.2,
524.2
Chioroethane
502.2, 524.2
Chloromethane
502.2, 524.2
o—Chlorotoluene
502.2, 524.2
p—Chlorotoluene
502.2, 524.2
Dibromomethane
502.2,
524.2
m—Dichlorobenzene
502.2, 524.2
Dichiorodifluoromethane
502.2,
524.2
1,1—Dichloroethane
502.2,
524.2
1,3—Dichloropropane
502.2, 524.2
2,2—Dichloropropane
502.2,
524.2
1,1—Dichioropropene
502.2,
524.2
1,3—Dichioropropene
502.2,
524.2
Fluorotrichioromethane
502.2, 524.2
Hexachiorobutadiene
502.2,
524.2
Isopropylbenzene
502.2,
524.2
p—Isopropyltoluene
502.2, 524.2
Naphthalene
502.2, 524.2
n—Propylbenzene
502.2, 524.2
1,1,2,2—Tetrachloroethane
502.2, 524.2
1,1,1,2—Tetrachloroethane
502.2,
524.2
1,2,3—Trichlorobenzene
502.2,
524.2
1,2,3—Trichloropropane
502.2,
524.2, 504.1
1,2,4-Trimethylbenzene
502.2, 524.2
1,3,5—Trimethylbenzene
502.2,
524.2
10
METHODS
FOR UNREGULATED
CONTAMINANTS
(CONT.)
2.
Analysis
for
the
13
unregulated
SOCs
listed
under
of
.141.40
shall
be
conducted
using
the
following.
methods.
Soc
Contaminants
Method
A1.dicarb
Aldicarb
sulfone
Aldicarb
sulfoxide
Aidrin
Butachior
C
arbaryl
Dicamba
Dieldrin
3—Hyd
roxycarbo
furan
Methomyl
Metol
achi Or
Metri
buzi
n
Propachl
or
531.1,
6610.
531.1,
6610
531.1,
6610
505,
508,
525.2,
508.1
507,
525.2
531.1,
6610
515.1,
515.2,
555
505,
508,
525.2,
508.1
531.1,
6610
531.1,
6610
507,
525.2,
508.1
507,
5252,
508.1
508,
525.2,
508.1
Other
mandatory
and
optional
procedures
for
conducting
analyses
of
unregulated
VOCs
and
SOCs
are
described
in
Sections
IV
and.V,
respectively,
of
this
Technical
Notes
document.
Sources.
for
EPA
Methods
502.2,
504.1,
505,
507,
508,
508.1,
515.1,
515.2,
524.2,
525.2,
531.1
and 551,
and
Standard
Method
6610
are
referenced
above
under
methods
for
organic
chemicals.
3.
Analysis
for
the
unregulated
inorganic
contaminant
listedunder
paragraph
(n)(12)
•of
§141.40
shall
be
conducted
Using
the
following
recommended
methods.
Contami
nant
Analytical’
Method
1
EPA
ASTM
SM
300.0
D4327—91
4110
375.2
0516—90
4500—S0
4
—F
4500—S0
4
—E
1.
Sources
for
the
Standard
Methods
and
ASTM
sulfate
inethodsare
referenced
above
under
methods
for
inorganic
chemicals.
The
EPA
methods
are
contained
in
“Methods
for
the
Determination
of
Inorganic
Substances
in
Environmental
Samples,”
EPA/600/R—93/100,
August
1993,
which
is
available
at
NTIS,
PB94—
121811.
paragraph
(n)
(11)
recommended
Sulfate
11
METHODS
FOR
FILTRATION
AND
DISINFECTION
1.
Microbiological,
pH,
and
Turbidity
Methods
To
comply
with
provisions
of
the
Surface
Water
Treatment
Rule
monitoring
under
Subpart
H
of
40
CFR
Part
141,
public
water
systems
must
conduct
analyses
of
total
coliforms,
fecal
coliforms,
heterotrophic
bacteria,
turbidity,
and
temperature
in
accordance
with
one
of
the
following
analytical
methods,
and
by
using
mandatory
procedures
for
turbidimeter
calibration,
which
are
specified
in
Section
IV
of
this
Technical
Notes
document.
Approved
methods
for
pH
are
described
above
under
“Methods
for
Inorganic
Contaminants.”
Organism
Methodology
Citation
1
Total
Coliforms
2
Total
Coliform
Fermentation
9221A, B,
C
Techni
que
3
’
4
’
5
Total
Coliform
Membrane
Filter
9222A,
B,
C
Technique
ONPG—MUG
Test
6
9223
Fecal
Coliforms
2
Fecal
Coliform
MPN
Procedure
7
9221E
Fecal
Coliform
Membrane
Filter
92220
Procedure
Heterotrophic
Pour
Plate
Method
9215B
bacteria
2
Turbidity
Nephelometric
Method
2130B
Nephelometric
Method
180.18
Great
Lakes
Instruments
Method
2
Temperature
2550
Footnotes
1
Except
where
noted,
all
methods
refer
to
the
18th
edition
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
1992,
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
2
The
time
from
sample
collection
to
initiation
of
analysis
may
not exceed
8
hours.
Lactose
broth,
as
commercially
available,
may
be
used
in
lieu
of
lauryl
tryptose
broth,
if
the
system
conducts
at
least
25
parallel
tests
between
this
medium
and
lauryl
tryptose
broth
using
the
water
normally
tested,
and
this
comparison
demonstrates
that
the
false—positive
rate
for
total
coliforms,
using
lactose
broth,
is
less
than
10%.
‘
Media
should
cover
inverted
tubes
at
least
one—half
to
two—thirds
after
the
sample
is
added.
No
requirement
exists
to
run
the
completed
phase
on
10
percent
of
all
total
coliforin—positive
confirmed
tubes.
6
The
ONPG—MUG
Test
is
also
known
as
the
Autoanalysis
Colilert
System.
12
A—I
Broth
may
be
held
up
to
3
months
in
a
tightly
closed
screwcap
tube
4CC.
at
8
“Methods
for
the
Determination
of
Inorganic
Substances
in
Environmental
Samples,”
EPA—600/R—93—100,
August
1993.
Available
at
NTIS,
P894—121811.
GLIMethod
2,
“Turbidity,”
November
2,
1992,
Great
Lakes
Instruments,
8855
Inc.,
North
55th
Street,
Milwaukee,.
Wisconsin
53223.
2.
Disinfectant
Residual
Methods
Public
water
systems
must
measure
residual
disinfectant
concentrations
with
one
of
the
analytical
methods
in
the
following
table.
The
methods
are
contained
in
the
18th
edition
of
Standard
Methods.
Corrections
tO
SM—4500—Cl—
E
and
4500—Cl—G,
and
procedures
for
conducting
continuous
measurements
of
chlorine
residuals
are
described
in
the
Technical
Notes
in
Section
IV
of
this
document.
Residual’
Methodology
Methods
Free
Chlorine
2
Aniperometric
Titration
4500—Cl
0
DPD
Ferrous
Titrirnetric
4500—Cl
F
DPD
Colorimetric
4500—Cl
G
Syringaldazine
(FACTS)
4500—Cl
I-I
Total
Chlorine
2
Amperomêtric
Titration
4500—Cl
D
Amperometric
Titration
4500—Cl
E
(low
level
measurement)
DPD
Ferrous
Titrimetric
4500—Cl
F
.
DPD
Colorimetric
4500—Cl
G
lodometric
Electrode
4500—Cl
I
Chlorine
Dioxide
Amperometric
Titration
4500—dO
2
C
DPD
Method
4500—Cl0
2
0
Amperometric
Titration
4500—dO
2
E
Ozone
Indigo
Method
4500—O.
B
Footnotes
1
if
approved
by
the
State,
residual
disinfectant
concentrations
for
free
chlorine
and
combined
chlorine
also
may
be
measured
by
usng
DPIJ
colorimetric
test
kits.
2
Free
and
total
chlorine
residuals
may
be
measured
continuously
by
a
adapting
specified
chlorine
residual
method
for
use
with
a
continuous
monitoring
instrument
provided
the
chemistry,
accuracy,
and
precision
of
the
measurement
remain
same.
Instruments
used
for
continuous
monitoring
must
be
calibrated
with
a
grab
sample
measurement
at
least
every
5
days,
or
with
a
protocol
approved
by
the
State.
13
SECTION
II.
METHODS
TO BE
WITHDRAWN
ON JULY
1,
1996
For
convenience
and
clarity,
the
methods to
be withdrawn
on
July
1,
1996
are
specified
in
this
document
in
lieu
of listing
them
in
the drinking
water
regulations
at
40 CFR Part
141.
The following
methods
may
be used
to obtain
certification
and
to
analyze
drinking
water compliance
samples
until
July
1,
1996.
However,
if
the
rule,
which
promulgates
this
withdrawal
action,
is
published
after
January
1, 1995,
the
withdrawal
date
becomes
18
months after
publication
of
the final
rule
in the
Federal
Register.
ANALYTICAL
METHODS
TO
BE
WITHDRAWN
FOR
INORGANIC
CONTAMINANTS
In
addition
to
methods
cited at
§141.23(k)(1),
the
methods
in the
following
table
only are
approved
until July
1,
1996
for analyses
for
antimony,
arsenic,
barium,
beryllium,
cadmium,
cyanide,
fluoride,
mercury,
nickel,
nitrate,
nitrite,
selenium,
sodium
and
thallium.
These
methods
were
previously
specified
at §141.23(k)(1),
except
arsenic,
fluoride
and sodium,
which
were
previously
specified
at §141.23(k)(2),
§141.23(k)(3)
and
§141.41(c),
respectively.
Antimony
6
Atomic
Absorption;
Furnace
204.2
Arsenic
4
Atomic
Absorption;
Furnace
206.2
Hydride—Atomic
Absorption
206.3
Spectrophotometric
206.4
D—2972—88A
Barium4
Atomic
Absorption;
Direct
208.1
Atomic
Absorption;
Furnace
208.2
Beryllium
4
Atomic
Absorption;
Furnace
210.2
Cadmium
6
Atomic
Absorption;
Furnace
213.2
Chromium
4
Atomic
Absorption;
Furnace
218.2
Manual
Distillation
followed
by
Spectrophotometri
c
Manual
Amenable,
Spectrophotometric
Fluoride
Manual
Distill.;
Color.
SPADNS
Manual
Electrode
Automated
Alizarin
Contaminant
Methodology
EPA
1
ASTM
2
SM
3
307B
Cyanide
335.2
335.1
340.1
340.2
340.3
14
0
Mercury
4
Manual,
Cold
Vapor
V
245.1
Nickel
4
Atomic
Absorption;
Direct
249.1
Atomic
Absorption;
Furnace
V
249.2
Nitrate
Manual
Cadmium
Reduction
353.3
Automated
Hydrazine
Reduction
353.1
Nitrite
Manual
Cadmium
Reduction
353.3
Spectrophotometric
V
354.1
Selenium
4
Atomic
Absorption;
Furnace
270.2’
Thallium
4
Atomic
Absorption;
Furnace
279.2
Sodium
Atomic
Absorption;
Direct
273.1
Atomic
Absorption;
Furnace
V
273.2
Flame
Photometric
V
D1428—64a
320A
Footnotes
V
V
V
V
1
“Methods
for
Chemical
Analysis
of
Water
and
Wastes,”
EPA—600/4—79—020,
March
1983.
Available
at
NTIS,
publication
order
number
PB84—128677.
V
.
2
Annual
Book
of
ASTM
Standards,
Part
31,
American
Society
for
Testing
and
Materials,
1916
Race
Street,
Philadelphia,
PA
19103.
Methods
320A and
307B
are
contained
in
the
14th
(1975)
and
16th
(1985)
editions,
respectively,
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
American
Public
Health
Association,
1015
Fifteenth
Street,
Washington,
D.C.
20005.
V
V
‘
Several
spectrochemical
techniques
are
approved
for
the
determination
of
metal
and metalloid
contaminants
in
drinking
water.
These
techniques
are:
inductively
V
coupled plasma—atomic
emission
spectrometry;
inductively
coupled
plasma—mass
spectrometry;
direct
aspiration
flame,
graphite
furnace,
and
platform
graphite
furnace
atomic
absorption
spectrometry.
To
conduct
these
measurements,
samples
must
not
be
filtered
prior
to
either
sample
digestion
or
“direct
analysis.”
V
Samples
are
acid preserved
with
nitric
acid
to
pH less
than
2,
held
for
16
hours,
and the
pH
verified
to
be
less
than
2
before
sample
processing
is
V
V
started.
In
addition,
the
turbidity
of
the
acidified
sample
must
be
measured
with
an
approved
method,
and
after
preservation
is
complete.
If
turbidity
is
greater
than
1
nephelonietric
turbidity
unit
(NTU),
sample
digestion
is
required
using
the
digestion
procedure
described
in
the
approved
method
(except
the
perchioric
acid
digestion
in
SM
3114B
must
not
be
used).
If the
acid
preserved
sample
contains
turbidity
less
than
1
NTU,
the
sample
may
be
analyzed
by
“direct
analysis”
without
digestion.
However,
irrespective
of
the
turbidity
of
the
sample,
when
determining
mercury
by
cold
vapor
atomic
absorption
(CVAA),
or
antimony,
arsenic,
or
selenium
(Sb,
As,
and
Se)
by
gaseous
hydride
atomic
V
absorption,
sample
aliquotsmust
be
digested
prior
to analysis.
Digestion
is
necessary,
because
organomercury
compounds
that
may be
present
in drinking
water
and
performance
samples
cannot
be
analyzed
by
CVAA
unless
converted
to
inorganic
15
mercury,
and
because
Sb,
As,
and
Se
each
must
be
converted
to
a
specific
valence
For
state
the
prior
determination
to
reduction
of
chromiumand
generation
by
graphite
of
the
furnace
hydride
analysis,
for
analysis.
an
appropriate
volume
of
30%
hydrogen
peroxide
(1—mL
of
30%
2
H
02
per
100
mL of
sample
or
standard) should
be
added
to
the
calibration
standards
and
the
sample
prior
to
analysis. The
addition
of hydrogen
peroxide
ensures
that
chromium
in
the
sample
and
calibration standards
is in
the
same
valence
state
fCr(III)).
This
provides
uniform
signal
response
in conventional
off—the—wall
graphite
furnace
determinations
of chromium.
Also,
calcium
concentrations
ranging from
10
to
50
mg/L
have
demonstrated
a
nonuniform
suppressive.
(less
than
20%)
matrix
effect
in
conventional
off—the—wall
nonpyrolytic
graphite
furnace
determinations
of
chromium.
If
calcium
is
present
at these
concentrations
in
the
chromium
sample,
use
of
the
matrix
modifier
magnesium nitrate
is
highly
recommended
(cf.
SM
3113A).
6
Thedistillatfon
procedure
in
EPA
Method
335.2
should
not
be
use,
and
the
sodium
hydroxide absorber
solution
final
concentration
must
be
adjusted
to 0.25
N
For
before
graphitecolorimetric
furnace
determinations
analysis.
of
selenium
when
nickel
nitrate
is used
as
the
matrix
modifier,
an
appropriate
volume
of
30%
hydrogen
peroxide
(2—mL
30%
H
202
per
100
niL of
sample
or
standard)
should
be
added
to both
the
calibration
standards and samples
prior
to
analysis.
It
has
been
demonstrated
that
the
addition
of
hydrogen
peroxide
enhances
the
absorption
signal
response
in
conventional
off—the—wall
graphite
furnace
determinations
of
selenium.
If
digestion
of
the
sample
is
required,
because
sample
turbidity
is
greater
than
1
NTU,
hydrogen
peroxide
is added
to
the
sample
at the
time
of
digestion.
Nickel
nitrate
(Ni
conc.
of
0.1%)
either
is
added
to
an aliquot
of
the
processed
sample
and
calibration standards
at the
time
of
analysis
or
may
be added
directly
in
the furnace (20
jg
Ni
per
20
jiL injection).
16
I,
ANALYTICAL METHODS
TO
BE
WITHDRAWN
FOR
LEAD,
COPPER,
AND
CORROSI.VITY
In
addition to the
methods
cited
at
§141.23(k)(1),
thie
methods
in
the
following
table
are
approved
until
July
1,
1996
for
analyses
for lead,
copper,
conductivity,
calcium, alkalinity,
orthophosphaté
and
silica.
These
methods
were
previously specified
on
June
30,
1994
(59
FR
33863)
at
§141.89(a)..
Contaminant
Methodology
EPA
1
Lead
2
Atomic
absorption;
furnace
technique
239.2
Copper
2
Atomic
absprption;
furnace
technique
220.2
Atomic
absorption;
direct
aspiration
220.1
Conductivity
Conductance
120.1
Calcium
2
EDTA
titrimetric
.
215.2
Atomic
absorption;
direct
aspiration
215.1
Alkalinity
Titrimetric
31.0.1
Orthophosphate
Colorimetric,
ascorbic
acid,
two
365.3
(Unfiltered,
reagent
.
no
digestion
Colorimetric.,
ascorbic
acid,
single
365.2
or
hydrolysis)
Silica
Colorimetric
370.1
Footnotes
1
“Methods for
Chemical
Analysis
of Water
and
Wastes,”
EPA—600/4—79—020,
March
2
1983.
Available
at
NTIS
as
PB84—128677.
To
conduct
these
measurements
samples
must
not
be
filtered
prior
to
either
sample
digestion or
“direct
analysis.”
Samples
are
acid
preserved
with
nitric
acid
to
pH
less
than
2,
held
for
16
hours,
and
the
pH
verified to
be
less
than
2
before
sample
processing is
started.
In
addition,
the
turbidityof
the
acidified sample
must
be
measured
using
an approved
method,
and
after
acid
preservation
is complete.
If
turbidity
is greater
than
1 nephelornetric
turbidity
unit
(NT1J),
sample
digestion
is
required
using
the
digestion
procedure described
in
the
approved method.
If
the
acid
preserved
sample
contains turbidity
less
than
1 NTU,
the
sample
may
be
analyzed
by
“direct
analysis”
without
digestion.
When
digestion
is
required,
the
total
recoverable
technique
as
defined
in
the
method
must
be
used.
17
ANALYTICAL
METHODS
TO
.BE
WITHDRAWN
FOR
ORGANIC
CONTAMINANTS
In
addition
to
methods
cited
at
§141.24(e),
the
methods
specified’
in
the
following
table
may
be
used
until
July
1,
1996
for
analysis
of
the
contaminants
specified
below.
Methods
502.1, 503.1
and
524.1
are
containedin
Methods
for
the
Determination
of
Organic
Compounds
in
Drinking
Water,
EPA/600/4—88/039,
December
1988,
Revised,
July
2991, which
is’
available
from
the
National
Technical
Information
Service
(NTIS),
PB91—231480,
U.S.
Department
of
Commerce,
5285
Port
Royal
Road,
Springfield,
Virginia
22161.
The
phone number is
800—553—6847.
Methods 501.1
and
501.2
for
analysis
of
total trihalomethanes
in
accordance
with
the
monitoring
requirements
specified
at
§141.30
will
be
printed
at
40
CFR
141.30,
Appendix
C
until
July
1,
1995.
Contaminant
EPA
Method
Benzene
503.1,
Carbon
tetrachloride
502.1,
Chi
orobenzene
502.
1,
1,2—Dichlorobenzene
502.2,
1,
4—Di
chl
orobenzene
502.
1,
1,2—Dichioroethane
502.1,
cis—Dichioroethylene
502.1,
trans—Di cM
oroethyl ene
502.
1,
Dichioromethane
502.1,
1,2—Dichloropropane
502.1,
Ethylbenzene
503.1,
Styrene
503.1,
Tetrachi
oroethyl
ene
502.
1,
1,1,1—Trichioroethane
502.1,
Trichloroethylene
502.1,
Toluene
503,1,
1,2,
4—Tn chl
orobenzene
503.1
1,
1—Di
chi
oroethyl
ene
502.1,
1,1,2—Tnichioroethane
502.1,
Vinyl chloride
502.1,
Xylenes
(total)
503.1,
Total Trihalomethanes
501.1,
§141.40(g),
EPA
Methods
502.1,
503.1
and
for
analysis
of
the
unregulated
VOC
40(e)
and
(j),
if
the
contaminant
is
method.
These
VOC
methods
are
contained
organic
contaminants,
524.1
524.1
503.1,
524.1
524.1
503.1,
524.1
524.1
524.1
524.1
524.1
524.1
524.1
524.1
503.1,
524.1
524.1
503.1,
524.1
524.1
524.1
524.1
524.1
524.1
501.2
ANALYTICAL
METHODS
TO
BE
WITHDRAWN
FOR
UNREGULATED
VOCS
In
addition
to
methods
cited
at
524.1
may
be
used until
July
1,
1996
contaminants
that
are
listed
in
§141.
listed
in
the
analytical
scope
of
the
in
the
EPA
manual described
above
for
I.
18
0
METHOD
TO’BE
WITHDRAWN
FOR
FILTRATION
AND
DISINFECTION
In
addition
to
methods
cited
at
§141.74(a)(5),
Standard
Method
408F
(Leuco
Crystal
Violet)
may
only
be
used
until
July
1,
1996
for analysis
of
free
chlorine
and
combined
chlorine
(chioramines).
This
method
is
contained
in
the
16th
edition
of
Standard
Methods
for the
Examination
of
Water
and
Wastewater,
1985,
American
Public
Health
Association,
10[5
Fifteenth
Street
NW,
Washington,D.C..20005.
19
SECTION
III.
RECOMMENDED
METHODS
FOR
SECONDARY
DRINKING
WATER
CONTAMINANTS
Analyses
of
aluminum,
chloride,
copper,
fluoride,
foaming
agents,
iron,
manganese,
odor,
silver,
sulfate, total
dissolved
solids
(TDS)
and
zinc
to
determine
compliance
under
§143.3
may be
conducted
with.the
methods
in
the
following
Table.
Criteria
for
analyzing
aluminum,
copper,
iron,
manganese,
silver,
and zinc
samples
with
digestion
or
directly
without
digestion,
and
other
mandatory
procedures
are
contained
in
the
Technical
Notes
in
Section
IV
of
this
document.
Measurement
of
pH
may
be
conducted
with
one
of
the
methods
listed
above
in
Section
I
under
“Methods
for
Inorganic
Chemicals.”
Contaminant
EPA
ASTM
1
SF1
2
Other
Aluminum
2OO.7
3120B
2OO.8
31138
2OO.9
3111D
Chloride
3OO.O
D4327—91
4110
4500—CY—D
Color
21208
Copper
2OO.7
D1688—90A
31208
2OO.8
D1688—90C
3111B
2OO.9
3113B
Fluoride
3OO.O
D4327—91
4110
129—71W
5
D1179—93A
4500F—B,D
380—75WE
5
D1179—938
4500F—C
4500F—E
Foaming
Agents
5540C
Iron
200.7
3120B
2O0.9
3111B
3113B
Manganese
200.7
3120B
200.8
3111B
200.9
31138
Odor
2150B
Silver
200.7
3120B
I372O856
2OO.8
3111B
20O.9
3113B
Sulfate
300.O
D4327—91
4110
375.2k
4500—S0
4
—F
4500—S0
4
—C,D
20
Contaminant
EPA
ASTM
1
SM
2
Other
TDS
2540C
Zinc
2O0.7
3120B
2O0.8
3111B
Footnotes
Annual
Book
of ASTM
Standards,
Vols.
11.01
and
11.02,
American
Society
for
Testing
and
Materials,
1916
Race
Street,
Philadelphia,
PA
19103.
2
18th
edition
of
Standard
Methods
for
the
Examination
of Water
and
Wastewater,
1992,
American
Public
Health
Association,
1015
Fifteenth
Street
NW, Washington,
D.C.
20005.
“Methods
for
the Determination
of
Metals
in
Environmental
Samples
—
Supplement
I,”
EPA—600/R—94—111,
May
1994.
Available
at
NTIS,
P894—184942.
‘
“Methods
for
the
Determination
of
Inorganic
Substances
in
Environmental
Samples,”
EPA—600/R—93—100,
August
1993.
Available
at
NTIS,
P894—121811.
Industrial
Method
No.
129—71W,
“Fluoride
in Water
and
Wastewater,”
December
1972,
and
Method
No. 380—75WE,
“Fluoride
in
Water
and
Wastewater,”
February
1976,
Technicon
Industrial
Systems,
Tarrytown,
NY
10591.
6
Available
from
Books
and Open—File
Reports
Section,
U.S.
Geological
Survey,
Federal
Center, Box
25425,
Denver,
CO
80225—0425.
21
SECTION
IV.
MANDATORY
METHOD
MODIFICATIONS
This section
contains
several
mandatory
method
modifications
in a
series
of Technical
Notes.
Each Technical
Note
is on
a
separate
sheet
to
allow
users
to remove
it, and
place
it with
the applicable
compliance
method(s).
The
parenthetical
number
(R),
which appears
adjacent
to
methodcitations
in
this
section,
refers
to the
publication
in
Section
VI
(References)
that
contains
the
referenced
method.
Tech. Notes
on
DW
Methods
October
1994
—
Section
IV
22
Mandatory
Method
Modifications
STANDARD
METHOD
(SM)
4500—C1—E
(R12),
CHLORINE
RESIDUALS
This
Technical
Note
corrects
a typographical
error
in SM
4500—Cl—E,
“Low
Level
Amperometric
Titration”
(R12).
This
method
is
currently
approved
at
§141.74(a)
for measurement
of
chlorine
residuals.
When
the
method
is
republished,
the
Standard
Methods
Committee
will
correct
an
error
1
in the
numerical
factor
in the
denominator
of
the
formula
in part
5
of the
method.
The
formula
is
on
page
+-43
of
the
18th edition
of
Standard
Methods.
The
correct
formula
must
have
a
factor
of 0.00564,
which
is
10
times
greater
than
the
factor
printed
in the
incorrect
formula.
1
Letter
from
Andrew
D..
Eaton,
“Error
in
4500—Cl
E,”
June
4,1993,
American
Public
Health
Association,
1015
Fifteenth
Street
NW, Washington,
D.C.
20005.
Tech.
Notes
on
OW
Methods
October
1994
— Section
IV
23
Mandatory
Method
Modifications
STANDARD
METHOD
(Sri)
4500—Cl—G
(R12),
CHLORINE
RESIDUALS
This
Technical
Note
recognizes
and corrects
an
error
in SM
4500—Cl—G
(R12).
This
DPD method
is
currently
approved
at
§141.74(a)
for
measurement
of
•chlorine
residuals.
The
method
as.
published
omits
instructions
that
would
allow
measurement
of
total
residual
chlorine
in drinking
water
samples.
The
Standard Methods
Committee
has deterniined
1
that an
editorial
omission,
not
a
technical
change,
occurred
in recent
versions
of
this
method.
The
error
will
be corrected
in
the
next
(19th)
edition
of
Standard
Methods.
The
simplified
procedure,
which
uses
DPD chemistry,
was
omitted
from
SM
4500—Cl—G
(18th
ed., para.
4,
p.
4—46).
EPA
corrects,
the,
Standard
Method
error,
by
printing
a
correction
to
paragraph
four below.
. The
correction
also
applies
to
the
16th
edition
version
of this
method,
SM 408E.
Simplified
Procedure
for
Total
Chlorine
“To obtain
monochloramine
and
dichioramine
together
as combined
chlorine
omit
step
4d
in
SM
4500—Cl-G
(monochioramine
determination).
To
obtain
total
chlorine
in
one
reading
add
the
full
amount
of potassium
iodide
at
the
start
with
the’
specified
amounts
of
buffer.
reagent
and
DPD
indicator.
Read
color
after
2
minutes..”
1
Letter
from
Andrew
D.
Eaton,
‘inquiry
on
Chlorine
Residual
4500—Cl
(18th
Edition),”
October
26, 1993,
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
Tech.
Notes
on
DW
Methods
October
1994
— Section
IV
24
Mandatory
Method
Modifications
PROTOCOL
FOR
CONTINUOUS
CHLORINE
RESIDUAL
MONITORING
In
this
Technical
Note
EPA
provides
specifications
for
continuous
monitoring
of
chlorine
residuals.
These
instructions
were
inadvertently
omitted
from the
Surface
Water
Treatment
Rule
(54
FR
27486,
June
29,
1989).
EPA
will
permit
a
grab
sample
method,
which
is
approved
for
chlorine
residual
monitoring at
§141.74(a), to
be
adapted
for
continuous
monitoring
of
free
or
total
chlorine
residuals
provided
the
chemistry,
accuracy,
and
precision
of
the method
are unchanged.
Instruments
used for
continuous
monitoring
must
be
calibrated
with
a
grab
sample
measurement
at
least
every
5
clays,
or
with
a
protocol
approved
by
the
State.
If
the
State
alsoapproves,
calibration
may
include
minor changes
in
the
reagent
mix
provided
the
overall
chemistry
of
the
method
is
not
changed.
Approved
grab
sampling
methods
for
chlorine
residual.
measurement
are
listed
below.
Free Chlorine
Ampe’ometric
Titration
4500—Cl
D
DPD
Ferrous
Titrimetric
.
4500—Cl
F
DPD
Colorimetric
4500—Cl
G
Syringaldazine
(FACTS)
4500—Cl
H
Total
Chlorine
Amperometric
Titration
4500—Cl
0
.
Amperometric
Titration
4500—Cl
E
(low
level
measurement)
DPD
Ferrous
Titrimetric
4500—Cl
F
DPD
Colorimetric
4500—Cl
G
todometric
Electrode
4500—Cl
I
1
If
approved
by
the
State,
residual
disinfectant
concenrations
for
free
chlorine
and
combined
chlorine
also
may
be
measured
by
using
DPD
colorimetric
test
kits.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
Mandatory
Method
Modifications
Residual’
Methodology
Methods
25
SPECTROP[IOTOMETRIC
DETERMINATIONS
OF
CYANIDE
Mandatory
Manual
Distillation
in
Cyanide
Methods
In
this
Technical
Note
EPA
emphasizes
that
spectrophotometric
measurements
of cyanide
in
water
samples
always
require
a
manual
digestion
of
the
sample
to
prepare
the sample
for
measurement
of
cyanide.
EPA
believes
emphasis
is
needed,
because
some
laboratories
seem
to
be
unaware
of
this
requirement.
All approved
spectrophotometric
methods
for
cyanide
are
specified
at 40
CFR
141.23(k)(1)
under
the
phrase,
“Manual
distillation
followed
by.”
Standard
Method
SM—4500—CN—C
(R12),
which
describes
the
mandatory
manual
distillation
procedure,
is
cited
in
the
rules
immediately
after
this
phrase.
Amenable”
spectrophotometric
methods
also
requiredistillation
prior
to
either
free
or total
cyanide
measurements.
The
approved
amenable,
manual
and
automated
spectrophotometric
methods
for
cyanide
are
ASTM
D2036—91B
and
D2036—
91A
CR11);
SM
4500—CN—F
and
4500—CN—G
(R12);
EPA
Methods
335.1,
335.2
and
335.3
CR14),
EPA
335.4
(R4);
and
USGS
1—3300—85
(R19).
(Note:
EPA
Methods
335.1
and
335.2
will
be
withdrawn
on
July
1,
1996,
and Method
335.3
has
been
replaced
by
Method
335.4).
To
avoid
manual
distillation,
laboratories
can
use
a
selective
electrode
method
for
cyanide,
which
is
discussed
below.
Selective
Electrode
Method,
SM
4500—CN—F,
(R12)
EPA
regulates
free,
not
total,
cyanide.
If
SM
4500—CN—F
is
used
to
determine
free
cyanide,
constant
ionic
strength
background
distillation
for
is
the
not
electrode
required.
measurement,
However,
to
samples
maintain
and
a
standards
must contain
the
same
concentration
of
sodium
hydroxide.
Reduced
Volume
Cyanide
Distillation
In
1994
EPA Method
335.3.was
replaced
with
Method
335.4.
The
technical
differences
between
the
methods
are
minor;
both
methods
require
manual
distillation
of
the
sample.
However,
EPA
improved
the
automation
of
procedures.
in.
Method
335.4,
and added
an
optional,
reduced
volume
distillation
procedure.
Method
335.4
does
not
contain
the
discussion
in
Method
335.3
of
an
alternate
ultraviolet
(UV) digestion
procedure,
because
approved
this
optional
UV
procedure,
and
because
EPA
believes
that
UY
digestion
will
underestimate
cyanide
concentrations
in
the
drinking
water
sample.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
26
Mandatory
Method
Modifications
In
this
Technical
Note,
EPA
is
approving
reduced
volume
distillation
for
all
spectrophotonietric
cyanide
methods.
Criteria
for
reduced
volume
distillation
are
as
follows.
“Reduction
in
digestion
or
distillation
volumes
is
&ceptable
provided
all
sample—to—reagent
ratios
are
maintained,
and
provided
the
final
sample
volume
is
sufficient
for
instrumental
measurement
of
cyanide.
Reduced
volume
considered
an acceptable
distillation
minor
apparatus,modification
when
employed
to
approvedas
described,
cyanide
can
be
methodology.”
EPA Method
335.2
(R14)
This
method
will
be
withdrawn
on
July
1, 1996.
ThisTechnical
Note
amends
Method
335.2
as follows.
The
sodium
hydroxide
absorber
solution
final
concentration
must
be
adjusted
to
0.25
N
before
colorimetric
analysis.
The
distillation
procedure
that
is
described
in
the
method
should
not
be
used,
because
it
uses
a
secondary
scrubber
that
does
not
work
well.
Tech.
Notes
on
DW
Methods
Octobór
1994
—
Section
IV
27
Mandatory
Method
Modifications
TURBIDIMETER
CALIBRATION
(R4,
R9,
R12)
EPA
Method
180.1
(R4), SM
21308
CR12)
and
GLI
Method
2
(R9)
are
approved
at
§141.74(a)
for
measurement
of
turbidity.
This
TechnicalNote
specifies
that
calibration
of
the
turbidimeter
must
be
made
either
by
the
use
of
a
formazin
standard
as
specified
in
the
approved
method
or
with
a
styrene
divinylbenzene
polymer
standard
(Amco
AEPA—1
Polymer).
This
reagent
is
commercially
available
from
Advance
Polymer
Systems,
Inc., 3696
Haven
Avenue,
Redwood
City,
California
94063.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
28
Mandatory
Method
Modifications
SAMPLE
DIGESTION
FOR
DETERMINATION
OF
METAL
CONTAMINANTS
This
Technical
Note
describes
when
and how
a
sample
must
be
digested
for
accurate
compliance
measurements
of
metals
in
drinking
water
samples.
Several
spectrochemical
techniques
are approved
for
the
determination
of
metal
and
metalloid
contaminants
in
drinking
water.
These
techniques
are:
inductively
coupled
plasma—atomic
emission
spectrometry;
inductively
coupled
plasma—mass
spectrometry;
direct
aspiration
flame,
graphite
furnace,
and
platform
graphite
furnace
atomic
absorption
spectrometry.
To
conduct
these
measurements,
samples
must
not
be
filtered
prior
to
either
sample
digestion
or
“direct
analysis.”
Samples
are
acid
preserved
with
nitric
acid
to
pH
less
than
2.
Preservation
is
complete
after
the
acidified
sample
has
been
held for
16
hours.
Before
sample
processing
is
started,
sample
pH
must
be
verified
to
be
less
than
2.
To
determine
whether digestion
of
the
sample
is
required,
the
turbidity
of
the
acidified
sample
must be
measured
using
an
approved
method
and
only
after
preservation
is
complete.
If
turbidity
is
greater
than
1
nephelometric
turbidity
unit (NTU),
sample
digestion
is
required
using
the
digestion
procedure
described
in
the
approved
method
(see
exception
below
for
SM
3114B).
If
the acid
preserved
sample
contains
turbidity
less
than
1
NTIJ,
the
sample
may
be
analyzed
by
“direct
analysis”
without
digestion.
However,
irrespective
of
the
turbidity
of
the
sample,
when
determining
mercury
by
cold
vapor atomic
absorption
(CVAA),
or
antimony
(Sb),
arsenic
(As)
or
selenium
(Se)
by
gaseous
hydride
atomic
absorption,
sample
aliquots
must
be
.
digested
prior to
analysis.
Digestion
of
the
sample,
which
is
described
in
the
applicable
method
1
,
is
necessary,
because
organomercury
compounds
that
may
be
present
in
drinking
water
and
performance
samples
cannot
be
analyzed
by
CVAA
unless
converted
to
inorganic
mercury,
and
because
Sb,
As,
and
Se
each
must
be
converted
to
a
specific
valence
state
prior
to
reduction
and
generation
of
the
hydride
for
analysis.
‘SM
31148
Exception
—
When
determining
arsenic
or
seleniumusing
gaseous
hydride
SM
31148
(R12), the
perchloric
acid
digestion
should
never
be
used.
See
the
Technical Note
on
“SM
3114B,
Arsenic
and
Selenium”
for
additional
instructions and explanations.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
29
Mandatory
Method
Modifications
STANDARD
METHOD
3114B (R12),
ARSENIC
AND SELENIUM
This
Technical
Note describes
an
important
safety
warning
when
using
sample
digestion
procedures
that are
described
in
SM 3114B
(R12).
Determination
of arsenic
and
selenium
by
gaseous
hydride,
atomic
absorption
requires
digestion
of
the sample
prior
to
analysis.
SM 3114B
describes
two
digestion
procedures.
One
procedure,
referred
to
as the
:“t0t
recoverable”
preparation,
uses
perchioric
acid
in
the
final
stage
of digestion.
This
perchioric
acid
digestion
procedure
is not
reciuired
by
EPA,
and should
be
avoided,
because
of
potential
danger
when
using
perchioric
acid,
and
because
a
special
fume hood
is
required.
When
using
method SM
3114B, the
digestion
procedure
described
in
paragraph
4.d,
Preparation
of samples
and standards
for
total
arsenic
and
selenium,
that specifies
the
use
sulfuric
acid
and
potassium
persulfate
should
be
utilized.
This
warning
is not
applicable
to
the ASTM
gaseous
hydride
methods
for
arsenic
and selenium,
because
the
methods
do
not
allow
use of
perchloric
acid
digestion.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
30
Mandatory’
Method
Modifications
ASTM
D3859—93B
(Ru)
AND STANDARD
METHOD
3113B
(R12),
SELENIUM
This
Technical
Note
concerns
graphite
furnace
determinations
of
selenium
with
ASTM
D3859—93B
CR11) or
SF1
31138
(R12).
When
nickel
nitrate
is
used
as
the
matrix
modifier,
an
appropriate
volume
of
30%
hydrogen
peroxide
(2-mL
30%
K
02
per
100 mL
of
sample
or
standard)
should
be
added
to
both
the
calibration
standards
and
samples
prior
to
analysis.
It
has
been
demonstrated
that
the
addition
of
hydrogen
peroxide
enhances
the
absorption
signal
response
in
conventional
off—the—wall
graphite
furnace
determinationsof
selenium..
If
digestion
of
the
sample
is
required,
because
sample
turbidity
is
greater
than
1
NTU,
hydrogen
peroxide
is
added
to
the
sample
at
the
time
of
digestion.
Nickel
nitrate
(Ni
conc.
of
0.1%)
either
is
added
to
an
aliiquot
of
the
processed
sample
and
calibration
standards
at
the
time
of
analysis
or
may
be
added
directly
in
the
furnace
(20
,g
Ni
per
20
,L
injection).
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
31
Mandatory
Method
Modifications
STANDARD METHOD
3113B
(R12),
CHROMIUM
This
Technical Note
describes
procedures
for
correctly
conducting
a
graphite
furnace determination
of
chromium
in
a
drinking
water
sample
using
SM
3113B
(R12). The
method
requires
that
an
appropriate
volume
of
30%
hydrogen
peroxide (1—mL
of
30%
H
202
per
100
mL
of
sample
or
standard)
be
added
to
the
calibration
standards
and
the
sample prior
to
analysis.
The
addition
of
hydrogen peroxide
ensures that
chromium
in
the
sample
and
calibration
standards is
in
the
same
valence
state,
chromium
[III].
This
provides
uniform
signal response
in
conventional
off—the—wall
graphite
furnace
determinations
of
chromium.
Calcium concentrations
ranging from
10
to
50
mg/L
have
demonstrated
a
nonuniform suppressive
(less
than
20%)
matrix
effect
in
conventional
off—the—
wall
nonpyrolytic
graphite
furnace determinations
of
chromium.
If calcium
is
present
at
these
concentrations
in
the
chromium
sample,
use
of
the
matrix
modifier
magnesium
nitrate
is
highly
recommended
(cf.
SM
3113A).
Tech.
Notes
on
DW Methods
October
1994
—
Section
IV
32
Mandatory
Method
Modifications
METHODS 502.2 (R16)
AND
524.2
(R3),
SORBENT
TRAPS
This
Technical
Note
describes
under
what
conditions
an
alternate
trap
may
be
used
in
EPA
Methods
502.2,
Rev.
2.0
(R16)
and
524.2,
Rev.
4.0.
(R3).
Both
methods
allow use
of
alternative
sorbents
to
trap
volatile
organic
compounds,
provided
all
quality
assurance
criteria
specified
in
the
method
are
met.
This
option
is
already included
in
Method
524.2
in
Sect.
:6.2.2,
but
an
explicit
requirement
not
to
change
other
method
conditions
is
missing.
EPA
notes
that
some
alternate
traps
may
not
work
under
Method
502.2
or
524.2
conditions,
because
the
purge
and
desorption
procedures
specified
in
the
methods
are
optimized
for
the
trap
media
specified
in
the
methods.
These
procedures
may
not
be
changed.
Specifically,
the
purge time,
purge
gas
flow
rate,
and
the
desorption
time
specified
in
the
method
may
not
be
changed,
because
EPA
has
no
data
to
show
that
reliable
or
reproducible
results
can
be
obtained
if
purging
or
desorption
times
or
flows
differ
from
the
specified
limits.
The
purging
and
desorption
conditions
for
these
methods
were
designed
to
achieve analytical
maximum efficiency.
The
purge
time
and
purge
gas
flow
rate
required
to
efficiently
purge the
target
analytes
from
the
water
sample
are
largely independent
of
the
sorbent
trapping
material.
Decreasing
the
purging
or
desorption
times
or
gas
flows
will
decrease
purging
efficiency
and/or
recovery
of
target analytes,
which will
have
a
negative
impact
on
method
precision.
Since
many of
the
potential
alternate
sorbents
may
be
thermally
stable
at
temperatures
higher
than
80°C,
alternate
traps
may
be
desorbed
and
baked
out
at
higher temperatures
than
those
described
in
the
current
method
revisions.
If
higher temperatures
are
used,
the
analyst
should monitor
the
data
for
analyte and
trap
decomposition.
This
Technical
Note
amends
Method
502.2,
Rev.
2.0
by
adding
the
following
sentence
to
the
end
of
Sect.
6.2.2.
“The
use
of
alternative
.sorbents
is
acceptable
provided
the
data
acquired
meets all
quality
control
criteria
described
in
Section
10,
and
provided
the
purge
and
desorption
procedures
specified
in
Section
11
of
the
method
are
not
changed.”
Method
524.2, Rev.
4.0
is
amended
by
changing
the
last
sentence
in
Sect.
6.2.2 to
read
as
follows.
“The
use
of
alternative
sorbents
is
acceptable
provided
the
data
acquired
meets
all
quality control
criteria
described
in
Section
9’,
and
provided
the
purge and
desorpt
ion
procedures
spe’cified
in
Section
11
of
the
method
are
not
changed.”
Tech.’Notes
on
DW
Methods
October
1994
— Section
IV
33
Mandatory
Method
Modifications
.
EPA
METHODS
502.2,
REV. 2.0
(R16),.
524.2,
REV.
4.0
(R3),
AND
551
(R15)
IN
SAMPLE ACIDIFICATION
This Technical
Note
clarifies
that
samples
must
be
acildified at
the
time
of
collection,
but after
they
have
been
dechlorinated.
Ac]dification
must
not
be
delayed
until the
samples
are received,
in
the
laboratory.
These
instructions
supersede
instructions
implied
or
explicit
that
may
be
contained
in
the methods.
•
Tech.
Notes
on
DW
Methods’
October
1994
Section
IV
34
Mandatory
Method
Modifications
METHOD
506
(R15),
ERRATA
IN
SUMMARY
This
Technical
Note
corrects
minor
errors
in
the
introductory
sections
of
Method
506
(R15),
and
emphasizes
that
clean
sodium
chloride
is
essential
to
an
accurate
analysis.
Method
506
is
used
to
determine
adipates
and
phthalates
in
drinking
water
samples.
The
summary
in
Section
2
of
Method
506
incorrectly
refers
to
use
of
a
ternary
solvent
mixture
to
conduct
the
liquid—liquid
extraction
of
the
sample;
the
correct
procedure
is
methylene
chloride
followed
by
hexane.
.
The
method
summary
also
omits
a
disk
elution
solvent.
Section
2
is
amended
to
correct
these
errors,
and
now
reads
in
entirety
as
follows.
“A
measured
volume
of
sample,
approximately
1—L,
is
extracted
with
methylene
chloride
followed
by
hexane
using
a
glass
separatory
funnel.
The
solvent
extract
is
isolated,
dried
and
concentrated
to
a
volume
of
mL
5
or
less.
The
extract
is
further
concentrated
by
using
a
gentle
stream
of
nitrogen
gas
to
reduce
the
sample
volume
to
1
mL
or
less.
Alternatively,
a
measured
volume
of
sample
is
extracted
with
a
liquid—
solid
extraction
(LSE)
cartridge
or
disk.
The
LSE
media
are
eluted
with
acetonitrile
followed
by
methylene
chloride
(disk
extraction)
or
with
methylene
chloride
only
(cartridge
extraction).
The
eluant
is
concentrated
using
a
gentle
stream
of
nitrogen
gas
or
clean
air
to
reduce
the
volume
to
1
mL
or
less.
The
analytes
in
the
extract
are
separated
by
means
of
capillary
gas
chromatography
using
temperature
programming.
The
chromatographically
separated
phthalate
and
adipate
esters
are
measured
with
a
photolonization
detector,
which
is
operating
at
10
eV,”
EPA
strongly
encourages
laboratories
to
clean
the
sodium
chloride
that
is
added
to
the
sample
by
carefully
following
the
heating
and
storage
instructions,
which
are
described
at
Sect.
7.5
of
the
method.
This
will
reduce
the
background
contaminatih
measured
in
the
laboratory
reagent
blank
samples.
Tech.
I1otes
on
DW
Methods
October
1994
—
Section
IV
35
Mandatory
Method
Modifications
IIETHOD
508
(R16),
DCPA
AND
HEXACHLOROCYCLOPENTADIENE
This
Technical
Note
approves
Method
508,
Rev.
3.0
(R16)
for
compliance
measurement
of
hexachiorocyclopentadiene,
provided
the
method
performance
criteria
specified
in
Section
9
of
Method
508.1
(R6)
are
met.
This
Note.
also
corrects
a
missing
entry
in
the
table
of
analytes
in
Sect.
1.1
of
Method
508;
the
CAS
Registry
number
for
DCPA
(dacthal)
is
1861—32—1.
Tech.
1otes
on
DW
Methods
October
1994
—
Section
IV
36
Mandatory
Method
Modifications
METHODS
515.1
(R16)
AND
515.2
(R3),
USE
OF
TMSD
This
Technical
Note
allows
and
describes
use
of
triniethylsilyl—
diazomethane
(TMSD)
as
an
alternative
derivatizing
reagent
in
Methods
515.1,
Rev.
4.0 (R16)
and
515.2,
Rev.
1.0
(R3).
EPA
is
approving
TMSD,
because
some
laboratories
prefer
not
to
use the
other
approved
derivatizing
reagent,
Diazald.
Since
TMSD increases
gas
chromatographic
background,
the
method
surrogate,
2,4—dichlorophenylacetic
acid,
cannot
be
used
at
concentrations
of
1
jg/L
or
lower.
Also,
Diazald,
not
TMSD,
must
be
used
if
dalapon
is
to
be
determined,
because
dalapon
is
not
amenable
to
esterification
with
TMSD.
If
dalapon
recovered
from
the
drinking
water
sample
is
incompletely
esterified,
dalapon concentrations
will
be
underestimated.
Laboratories
wishing
to
avoid
use
of
Diazald
may use
Method
552.1
to
determine
dalapon,
and
Method
515.1
or
515.2
or
555
for the
other
chlorinated
acid
herbicides.
Steps,
which
replace
or
augment
the
calibration
and
extract
esterification
(Sect.
11.4)
method
descriptions
when
TMSD
is
used,
are
dQscribed
below.
The following
procedure
was
written
for
Method
515.2,
which
uses liquid—solid
extraction
(LSE).
Analysts
using
TMSD
with
liquid—liquid
extraction
(LLE)
Method
515.1
should
omit
steps
specific
to
LSE,
and
include
appropriate
LLE steps
from
Method
515.1.
In
particular,
the
amounts
of
TMSD,
acetic
acid,
and internal
standards
to
be
added
may have
to
be
adjusted
when
the
TMSD
procedure
is
adapted
for
use
with
Method
515.1.
These
adjustments
may be
necessary,
if
the concentration
ratio
of
original
sample
to
final
extract
is
different
in
the two
methods.
USE
OF
TRIMETHYLSILYLDIAZOMETHANE
TO
ESTER
1FY
ACID
HERBICIDES
IN
METHOD
515.21,2
1.
INTRODUCTION
Trirnethylsilyldiazomethane
(TMSD)
is
available
from
acoinmercial
supplier
(currently
the
Aldrich
Chemical
Company
is
the
sole
supplier)
as
a
2
molar
solution
in
hexane.
TMSD
is
stable
during
storagein
this
solution.
It
should
be
noted
that
the
gas
chromatographic
background
is
somewhat
increased
when
TMSD
is
used
as
the
derivatizing
reagent
instead
of
the
generated
diazomethane.
Although
no
method
analyte
is
affected
by
this
increased
background,
the
recommended
surrogate,
2,4—dichloro—
phenylacetic
acid,
is
masked
by
an
interfering
peak.
This
renders
the
surrogate
useless
at
1
tg/L or
lower.
Any
compound
found
suitable
when
TMSD
is
used
is
acceptable
as
a
surrogate.
Trimethylsilyldiazomethane
can
be
used
to
efficientlymethylate
the
following
acid
herbicides:
Tech.
•Notes
on
DW
Methods
October
1994
—
Section
IV
37
Mandatory
1ethod
Modifications
Chemical
CAS
Registry
Nurnr
Acifluorofen
50594—66—6
Bentazon
25057—89—0
Chioramben
133—90—4
Dacthal.
1861—32—1
Dicaniba
1918—00—9
Dichiorprop
120—36—5
Dinoseb
88—85—7
3,5—Dichlorobenzoic
acid
51—36—5
2,4—D
94—75—7
2,4—DB
94—82-6
5—Hydroxydicamba
7600—50—2
Pentachi
orophenol
87—86—5
Picloram
1918—02—1
2,4,5—TI’
(Silvex)
93—72—1
2,4,5—T
93—76—5
TMSD
may
not
be used
to
esterify
dalapon.
The following
procedures
to
methylate
the
herbicides
mast
be
followed.
2.
CALIBRATION
OF
THE
GAS
CHROMATOGRAPH/ELECTRON
CAPTURE
I)ETECTION
(GC/ECD)
SYSTEM
Calibrate
the GC/ECD
system
using
fortified
reagent
water
samples,
and
use two
sets
of
calibration
solutions
to
prevent
coelution.
The
presence
of
coeluting
analytes
makes
confirmation
of
positives
mandatory
before
taking
action
on
a
result.
Follow
the
procedure
described
below
using
TMSD to
niethylate
the
herbicides.
Five
concentration
levels
are
recommended.
3.
PROCEDURE
Carry
out
the
hydrolysis,
clean—up,
and
extraction
of
the
method
analytes
as described
in
Method
515.2
up
to
Sect.
11.2.4,
or
inMethod
515.1
up
to
Sect.
11.4.
Users
of Method
515.1
should
begin
below
where
the
2
M
TMSD
solution
is
added.
Elute
the
herbicides
from
the
disk
by
passing
two
2
mLaliquots
of
methyl
tertiary
butyl
ether
(MTBE)
through
the
disk
into
the
collection
tube.
Rinse
the
sample
container
with
4
niL
of
MTBE
and
pass
it
through
the
disk
into
the
tube.
Transfer
the
MTBE
extract
from
the
collection
tube
intal
an
anhydrous
sodium
sulfate
drying
tube
which
has
been
pre—wetted
with
I
niL
MTBE.
Be
sure
to
discard
any water
layer.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
IV
38
Mandatory
Method
Modifications
Before
the
extract
passes
completely
through
the
sodium
sulfate,
add
an
additional
2
mL
of
MTBE
as
a
rinse.
Concentrate
the
dried
extract
to
approximately
4
mL.
Add
methanol
(approx.
1
niL)
to
the
extract
to
yield
a
20%
(v/v)
methanol
in
MTBE
solution.
Adjust
the
volume
to
5
niL
with
MTBE.
(TMSD
produces
the
most
efficient
methylation
of
the
herbicides
in
a
20%
methanol,
80%
MTBE
solution.)
Add
50
jiL
of
the
2
N
TMSD
solution
to
each
5
mL
sample
extract.
(Verify
this
volume
if
Method
515.1
is
used.)
Place
the
tube
containing
the
extract
into
a
heating
block
at
50°C
and
heat
the
extract
for
1
hour.
Allow
the
extract
to
cool
to
room
temperature,
then
add
100
#L
of
2
N
acetic
acid
in
methanol
to
react
any
excess
TMSD.
(Verify
this
volume
if
Method
515.1
is
used.)
Fortify
the
extract
with
100
itL
of
the
internal
standardsolution
(Method
515.2,
Sect.
7.17;
Method
515.1,
Sect.
7.19)
to
yield
a
concentration
of
0.020
g/mL.
(Verify
this
if
Method
515.1
is
used.),
Proceed
with
the
identification
and
measurement
of
the
analytes
using
GC/ECD
according
to
the
procedures
described
in
the
method.
1
“Use
of
Trimethylsilyld’iazomethane
as
a
Substitute
Reagent
for
the
Esterification
of
Phenoxy
Herbicides,”
J.
Collins
and
W.J.
Bashe,
Technology
Applications,
Inc.,
July
27,
1993
[Project
performed
under
EPA
Contract
68-Cl—
0022,
J.W.
Eichelberger,
Work
Assignment
Manager]
2
Amounts
of
TMSD,
acetic
acid,
internal
standards
and
other
reagents
may
have
to
be
adjusted
when
the
TMSD
procedure
is
adapted
for
use
with
Method
515.1.
These
adjustments
will
be
necessary,
if
the
concentration
ratio
of
original
sample
to
final
extract
is
different
in
the
two
methods.
Tech.
Notes
on
OW
Methods
October
1994
—
Section
IV
39
Mandatory
Method
Modifications
METHOD
524.2,
REV.
4.0
(R3)
QUALITY
ASSURANCE,
VOC
DATA
.
This
Technical
Note
corrects
or
clarifies
quality
assurance
steps
in
Method
524.2,
Rev.
4.0
(R3),
and
provides
data
for
two
VOCs
that
was
omitted
•
in
the
published
method.
Changes
in
Quality
Assurance
Procedures
EPA
is
changing
some
instruttions
in
Sections
9
(quality
control)
and
10
(calibration)
of
Method
524.2
that
may
be
conflicting
or
confusing.
The
changes
described
in
this
Note
also
apply
to
Method
502.2,
Rev.
2.0
(R16) to
the
extent that
the
same
problems
are
in
the
quality
control
(Section
10)
and
calibration (Section
9).
Section
9.3, Initial
Demonstration
of
Accuracy
——
EPA
has
been asked
to
make the
accuracy
criteria
(±20%),
which
are
part
of
an
initial
demonstration
of
capability
(IDC),
in
Sect.
9.3.3
of
Method
524.2
the
same
as
the
accuracy
criteria
(±30%).
in
the
section
on
continuing
calibration
checks
(Sect.
10.3.5).
These
criteria
will
not
be
changed.
EPA
specified
different
criteria,
because
the
IDC
and
Continuing
Calibration
measurements
are
evaluating
different
controls.
EPA
believes
the
IDC
measurement,
which requires
analysis
of
a
series
of
laboratory
fortified
blanks,
should
be
more
accurate
than
the
Continuing
Calibration
measurement.
To
explain
this
difference
in
accuracy
criteria,
and
to
remove
an
incomplete
reference
to
the
SDWA,
Sect.
9.3.3
is
revised
in
this
Note,
Section
9.3.3
is
superseded
in
its
entirety
as
follows:
“Some
analytes,
particularly
early
elating
gases
and
late
elating
higher
molecular
weight compounds,
will
be
measured
with
less
accuracy
and
precision
than other
analytes.
However,
the
accuracy
and
precision
for
all
analytes
must
fall
within
the
limits
expressed
below.
If
these
criteria
are
not
met
for
an
analyte
of
interest,
take
remedial
action
and
repeat the
measurements
for
that
analyte
until
satisfactory
performance
is
achieved.
For
each analyte,
the
mean
accuracy
must
be
80—120%
(i.e.
an
accuracy
of
±
20%).
The
precision
of
the
recovery
(accuracy)
for
each
analyte
must
be
less
than twenty
percent
(<20%).
These
criteria
are
different
than the
±
30%
response
factor
criteria
speâified
in
Sect.
10.3.5.
The criteria
differ,
because
the
measurements
in
Sect.
9.3.3
as
part
of
the
initial
demonstration
of
capability
should
be
more
stringent
than
the
continuing
calibration
measurements
in
Sect.
10.3.5.”
Section 9.6
LFB
Criteria
——
This
step
in
Method 524.4
requires
a
single
laboratory
fortified
blank
(LFB)
to
be
measured
with
each
batch
of
samples,
and
with
a,n
accuracy
that
is
specified
in
Sect. 9.3.3
(i.e.
±20%),
whereas
Sect.
10.3.5
requires
the
same
sample
be
analyzed
with
an
accuracy
of
±30%.
EPA
is
removing
this
conflict
by
changing
the
accuracy
requirement
to
be
±30%
in
Sect.
9.6.
Tech.
‘Notes
on
DW
Methods
October
1994
—
Section
IV
40
Mandatory
1ethod
Modifications
Section
9.6 is
superseded
in
its
entirety
as
follows:
“Use
the procedures
and
criteria
in
Sects.
10.3.4
and
10.3.5
to
evaluate
the
accuracy
of
the measurement
of
the
laboratory
fortified
blank
(LFB),
which
must
be
analyzed
with
each
batch
of
samples
that
is
processed
as
a
group
within
a
work
shift.
If
more
than
20
samples
arein
a
work
shift
batch,
analyze
one
LFB
per 20
samples.
Prepare
the
LFB
with
the
concentration
of
each analyte
that
was
used
in the
Sect.
9.3.3
analysis.
If
the acceptable
accuracy
for
this
measurement
(±30%)
is
not
achieved,
the problem
must
be
solved
before
additional
samples
may be
reliably
analyzed.
Since
the calibration
check
sample
in
Sect.
10.3:5
and
the
LFB
are
made
the same
way and since
procedural
standards
are
used,,
the
sample
analyzed
here
may
also
be
used
as
the
calibration
check
in
Sect.
10.3.5.
Add
the
results
of
the
LFB
analysis
to
the
control
charts
to
document
data
quality.”
Section
9.5
LRB Analysis
——
This
step
in
Method
524.2
states
that
a
field
reagent
blank
may
be
used
in
lieu
of
a
laboratory
reagent
blank
(LRB).
This
is
not correct.
An
LRB
must always
be
analyzed
with
each
batch
(as
defined
at
Sec:t.
9.6)
of
20
samples.
This
Note amends
Sect.
9.5
by
deleting
the
erroneous
second
sentence.
Section
9.5
is
superseded
in
its
entirety
as
follows
“LABORATORY
REAGENT
BLANKS
(LRB)
--
With
each
batch
of
samples
processed.
as
a
group
within
awork
shift,
analyze
a
LRB to
determine
the
background
system
contamination.”
Section
9.7
FRB Analysis
——
This
step
in
Method
524.2
states
that
a
“field
reagent
blank
should
be
analyzed”
with
each set
of samples.
This
may
cause
unnecessary
work.
A
field
reagent
blank
is
collected
as
a
precaution
against
false
positive
results
that
may occur
if
the sample
is
contaminated
in
the
field.
Thus,
a
field
reagent
blank
analysis is
only
required
when
contamination
is
detected
in the
compliance
sample.
This Note
clarifies
when
the
samples
must
be
analyzed
by
ameiding the
first
sentence
in
Sect.
9.7.
Section
9.7
is
superseded
in
its
entirety
as
follows:
“If a
water
sample
is
contaminated
with
an
analyte,
verify
that
it
is
not
a
sampling
error
by
analyzing
a
field
reagent
blank.
The
results
of
these
analyses
will
help
define
contamination
resulting
from
field
sampling,
storage
and transportation
activities.
If
the
field
reagent
blank
shows
unacceptable
contamination,
the
analyst
should
identify
and
eliminate
the
contamination.”
Tech.
Notes
on DW
Methods
October
1994
—
Section
IV
41
Mandatory
Nethod
Modifications
Sctior
10,
Calibration
——
There
can
be
a
conflict
between
the
instructions
in
Sect.
9.6
in
Method
524.2,
which
define
a
batch
as 20
samples,
and
Sect.
10.1,
which
requires
calibration
every
8
hours.
Since
a
typical
chromatographic
run
exceeds
35
minutes,
20
samples
are
measured
in
about
11,
not
8,
hours.
This
Note
removes
the
potential
conflict
by
explaining
when
calibration
must
be
checked.
Section
10.1
is
superseded
in
its
entirety
as
follows:
“Demonstration
and
documentation
of
acceptable
initial
calibration
is
required
before
any
samples
are
analyzed.
In
addition,
acceptable
performance
must
be
confirmed
intermittently
throughout
analysis
of
samples
by
performing
continuing
calibration
checks.
These
checks
are
required
at.the
beginning
of
each
work
shift,
but
no
less
than
every
12
hours.
Additional
periodic
calibration
checks
are
good
laboratory
practice.
Since
this
method
uses
procedural
standards,
the
analysis
of
the
laboratory
fortified
blank,
which
is
required
in
Sect.
9.6,
may
be
used
here
as
the
calibration
check
sample.”
Tech.Notes
on
OW
Methods
October
1994
—
Section
IV
42
Mandatory
Method
Modifications
Performance
Data
for
cis—and---trans
1,3—dichioropropene
Tech.
Notes
on DW
Methods
October
1994
— Section
IV
Mandatory
Method
Modifications
,
EPA
omitted
performance
data
for
two
unregulated
VOCs,
cis—
1,3—dichioropropene
and
trans—1,3—dichloropropene.
The
following
table
replaces
Table
7
in Method 524.2,
Rev.
4.0.
TABLE
7.
ACCURACY AND
PRECISION
DATA
FROM
SEVEN
DETERMINATIONS
OF METHOD
ANALYTES
IN
REAGENT
WATER
USING
WIDE
BORE
CAPILLARY
COLUMN
NUMBER
4
.
Mean
Rel.
Method
True
Conc.
Std.
Detect.
Conc.
Detected
Dev.
Limit
Compound
(pg/L)
(ug/L)
(%)
(J.LgIL)
Acetone
1.0
1.6
5.7
:
0.28
Acrylonitrile
1.0
0.81
8.7
0.22
Allyl
chloride
1.0
0.90
4.7
0.13
2—Butanone
2.0
2.7
5.6
0.48
Carbon
disulfide
0.20
0.19
15
0.093
Chloroacetonitrile
1.0
0.83
4.7
0.12
1—Chiorobutane
1.0
0.87
6.6
0.18
t—Dichloro—2—butene
1.0
1.3
8.7
0.36
1,1—Dichioropropanone
5.0
4.2
7.7
1.0
c—1,3—Dichloropropene
0.20
0.20
3.1
0.020
t—1,3—Dichloropropene
0.10
0.11
14
0.048
•
Diethyl ether
1.0
0.92
9.5
0.28
Ethyl
niethacrylate
0.20
0.23
3.9
:
0.028
Hexachloroethane
0.20
0.18
10
0.057
2—Hexanone
..
1.0
1.1
12
0.39
Methacrylonitrile
1.0
0.92
4.2
0.12
Methylacrylate
1.0
1.2
12
0.45
Methyl
iodide
0.20
0.19
3.1
0.019
Methylmethacrylate
1.0
1.0
13
0.43
4—Methyl—2—pentanone
0.40
0.56
9.7
0.17
Methyl—tert—butylether
0.40
0.52
5.6
0.090
Nitrobenzene
2.0
2.1
18
1.2
2—Nitropropane
1.0
0.83
6.2
0.16
Pentachloroethane
0.20
0.23
20
0.14
Propionitrile
1.0
0.87
5.3
0.14
Tetrahydrofuran
5.0
3.9
13
1.6
43
EPA
METHOD
531.1
(R16)
AND
SM 6610
(RB),
STORAGE
OF
SAMPLES
This
Technical
Note removes
the requirement
in
Methods
531.1,
Rev.
3.0
(R16) and
SM 6610
(RB)
to
freeze the
samples.
Sect.
8.2.4
of
Method
531.1
requires
buffered
samples
to
be stored
at
minus 10°C.
EPA
realizes
that
this
is impractical
and unnecessary.
After
reviewing
time
storage data,
EPA
concluded
that
samples
buffered
to a pH
of 3
or less
may
be
stored
at 4°C.
-
The
data supporting
this conclusion
is contained
in
Table 6610:11
of
SM
6610.
To
reflect
this change
this
Note
supersedes
Sect.
8.2.4
of
EPA Method
531.1 in
its
entirety
as follows.
Users of
the
Standard
Method
should
make
appropriate
changes
to the
procedures,
which are
described
in
Paragraph
2
(Sampling
and
Storage)
of SM
6610.
“Samples
must
be iced
or refrigerated
at
4°C from
tie
of collection
until
analysis
is
begun.
Although,
preservation
study
results
of
up
to
28
days indicate
method
analytes
are
not
labile
in
water
samples
when
sample pH
is adjusted
to
3 or less,
and samples
are
shipped
and stored
at
4°C, analyte
lability
may
be
affected
by
the matrix.
Therefore,
the
analyst must
verify
that the
preservation
technique
is. applicable
to the
samples
under
study.”
Tech. Notes
on
DW Methods
October 1994
—
Section
IV
44
Mandatory
Method
Modifications
METHOD
551
(R15),
PENTANE
•
This
Technical
Note
allows
optional
use of
pentane
as the
extraction
solvent
for
some of
the
analytes
in
EPA Method
551
(R15).
Since
a
change
in
the extraction solvent
in
any
method
is
a
change
in
the
chemistry
of the
method,
an
alternative
solvent
must
be validated
and
approved
by
EPA
for
each
method
analyte. EPA
has
approved
only
methyl
t—butyl
ether
(FITBE)
and pentane
for
use as
extraction
solvents
in
Method
55]..
Pentane
may not
be
used
to
extract
chioral
hydrate;
MTBE
is
approved
for
all
Method
551
analytes.
Tech.
Notes
on
DW
Methods
October
1994
— Section
IV
45
Mandatory
Method
Modifications
EPA
METHOD
549.1
(R3),
SAMPLE
CONTAINERS
This
Technical Note
clarifies:
that
the
amber
sample
bottle
specified
in
Section
6
(Equipment and
Supplies)
of
Method
549.1,
Rev.
1.0
(R3), can
be
made
of
any
type
of
plastic.
The
bottle
does
not
have
to
be
PVC
as
stated
in
the
method.
Tech.
Notes
on
DW
Methods
October
1994
— Section
IV
46
Mandatory
Method
Modifications
ALTERNATIVE
LIQUID—SOLID
EXTRACTION
CARTRIDGES
AND
DISKS
This
Technical
Note
provides
criteria
for
judging
the
equivalency
of
liquid—solid.extraction
(LSE) cartridges
and
disks
for
use
in methods
that
allow
use
of
LSE
technology.
This
Note
supersedes
the
phrase
“or
equivalent”
that
is
used
in
some
methods to
describe
selection
of
alternative
LSE
cartridges
or
disks.
Although
EPA
welcomes
innovative
LSE
technology,
EPA
will
not
approve technology
that
compromises
the
reliability
of
the
analysis.
Liquid—solid
extraction
is
performed
using
various
sdrbents
that
are
either
packed
into
a
cartridge or
enmeshed
in
a
disk
of
inert
support
material. EPA
methods describe
the
cartridge
or
disk
that
was
used
to
develop
the
LS.E
procedure,
and
to produce
the
data
which
is published
in
the
method.
If
a
product
is
mentioned
in
the
methods,
it
is
for
information
purposes
only.
EPA
believes various
LSE
cartridges
and
disks
may
be
used,
provided
they
meet
all
quality control requirements
of
the
method,
and
provided
they
contain
a
sorbent
that
uses
the
same
physicochemical
principles
as
the
cartridge
or
disk
that
is
described
in
the
approved
LSE
method.
To
denionstrate
that
alternative
LSE
cartridges
and
disks
meet
all
quality
control
criteria, the
analyst
must
be
aware
of the
chemistry
of
the
method.
For
example,
in
evaluating
Method 552.1
the
recovery
of
the
free
acid
(not
a
chemical
derivative)
from
the
water
sample
must
be
tested
with
the
alternative
LSE
cartridge
or
disk.
In
judging
LSE
disk
media,
both
the
sorbent
and
the
support
must
be
evaluated.
In
the
case
of
sorbents,
similarities
in
polarity
are
not
sufficient.
For
example,
a
C
18
—Silica
sorbent
may
not
perform
the
same
as
a
,
styrene
divinylbenzene copolymer
sorbent.
Thus,
these
sorbents
would
not
be
considered to
be
equivalent.
In
judging
supports,
any
physical
support
used
to
hold
the
sorbent
is
acceptable
provided
the
support
is
inert
and
compatible
with
the
solutions
or
solvents
required
in
the
conditioning
and
elution
steps
of
the
method. However,
any
sorbent
conditioning
or
elution
steps,
which
are
specified
in
the
method
must
not
be
modified
or
eliminated
to
accommodate
the
support
material.
For
example,
Method
552.1
was
developed
and
validated
with
ion
exchange cartridges
to
determine
dalapon
and
haloacetic
acids.
To
efficiently
extract
the
acids, the
ion
exchange
resin
must
be activated
with
a
sodium
hydroxide
rinse.
In
judging
the
equivalency
of
an
alternative
disk
EPA
would
still
require the
rinse,
because
EPA
has
no
data
to
support
making
the
rinse
optional.
Tech.
Notes
on
DW
Methods
October
1994
— Section
IV
47
Mandatory
Method
Modifications
SECTION
V.
RECOMMENDED
METHOD
MODIFICATIONS
This
section
contains
several
optional
procedures
and
recommended
modifications
to
compliance
methods.
Each
optional
or
recommended
procedure
is
on
a
separate
page
to allow
users
to
remove
it,
and
place
it
with
the
applicable method(s). The
parenthetical
number
(R),
which
appears
adjacent
to
method
citations in this
section,
refers
to
the
publication
in
Section
VI
(References) that
contains
the
referenced
method.
Tech.
Notes
on DW
Methods
October
1994
—
Section
V
48
Recommended
Method
Modifications
METHOD
100.1
(RiB),
ASBESTOS
GUIDANCE
•
This
Technical
Note
does
not
change
Method
100.1
(R18).
It
describes
how
to
make
some
steps
in
the method
specifically
applicable
to
the
drinking
water
standard
of
asbestos
fibers
greater
than
10
pm in
length.
This
guidance
is
needed
because
the
asbestos
method
was
not
designed
specifically
for
measuring
fibers
greater
than
10 pm
in
length,
and
because
laboratories
may
not
wish
to
use
an
ozone/UV
generator
to
prepare
the
samplefor
analysis.
EPA
METHOD
100.1
DETERMINATION OF ASBESTOS
FIBERS
IN WATER
OGWDW
GUIDANCE
AND
CLARIFICATION
FOR DRINKING
WATER
1.
Approximately 800
mL
of sample
should
be
taken
in
1—L
bottles.
Glass
sampling
bottles
are
preferable
to
plastic.
If
plastic
bottles
are
used,
polyethyleneis
better
than
polypropylene.
Do
not
use
acid
or
mercuric
chloride
as
preservatives.
•Before
collecting
the
sample,
the
water
must
be
allowed
to
run until
the
temperature
has
stabilized,
indicating
that
the
water
is
representative
of the
main
water
line.
Samples
must
be taken
in duplicate.
Store
samples
in the
dark
at
4°C.
2.
To
avoid
use
of the
ozone,
ultraviolet
(UV) generator,
samples
must
be filtered
on the
‘polycarbonate
(PC)
filter
in
the
laboratory
within
48
hours
of
collection.
If
the
holding
time
is
exceeded,
the
sample
must
be
treated
to break
down
microbiological
contaminants.
This
is
done
immediately
prior
to
filtration
by treating
the
sample
in the
original
container
with
ozone,
UV—light,
and
resonicating
it
to
disperse
the
fibers.
3.
Up
to
5 samples
may
be
composited.
Sample
compositing
must
be done
in
the
laboratory
on
samples
which
are less
than
48—hours—old
or
have
been
individually
ozone/UV
treated
in
their
original
sample
containers. Samples’must
be
sonicated
and
equal
amounts
withdrawn
to
make
up
the
composite.
It may
also
be
prudent
to
filter
an
aliquot
of
each
individual
sample
for
analysis
in.
case the
composite
sample
exceeds
1/5
of the
MCL
(1.4
NFL
>10 jim
long).
If
this
is
not
done,
the original samples
can
only
be
filtered
if
they
are less
than 48—hours—old
and have
been
resonicated
or
have
been
retreated
with
ozone—UV
and resonicated.
4.
Only
0.1 jim
pore
size
PC filter
membranes
may
be
used.
Filters
must
be
taken
from
a lot
which
has been
prescreened
for background
contamination.
This
is
particularly
important
if
fibers
less
than
10
jim
are to
be
counted
because
PC
filters
may
be
contaminated
with
asbestos
fibers
shorter
than
10 pm.
The
PC
filter’
must
be
backed
by
a
methyl
cellulose
ester
(MCE)
filter
to diffuse
the
vacuum
across
the
membrane.
Use
5
jim
pore
size
MCE
membrane
as the
backing
filter.
Tech.
Notes
on
DW Methods
October
1994
— Section
V
49
Recommended
Method
Modifications
5.
A
filtration
apparatus
with
straight
vertical
sides
is
preferred
to
one with
tapered
sides
to
avoid
loss
of
fibers
settling
on
tapered
sides
of
the
funnel.
6.
States
agencies
may choose
to
require
the
counting
offibers
•less
than
10
pm long
to
help
judge
the
condition
of
asbestos/cement
pipes.
Certification
lists
must
identify
whether
labs
count
all
fibers
or
only
those
over
10
pm,
and whether
the
lab
is
certified
by
a
state
or
EPA
region.
7.
A
calibrated
magnification
of
at
least
10,000X
±%
is
adequate
for
counting
fibers
over
10
pm in
length.
A
minimum
spot
size
of
250
nm
or
smaller
is
required
for
this
analysis.
8.
For compliance
analysis
of
asbestos
in
drinking
water
samples,
an
analytical
sensitivity
200,000
fibers
per
liter
(0.2MFL)
is
required,
subject
to
the
following
stopping
rules:
a.
Analysis
may
be
terminated
at
the
completion
of
the
grid
opening
during
examination
of
which
an
analytical
sensitivity
of
0.2MFL
is
achieved,
or
at
the
completion
of
the
grid
opening
which
contains
the
100
th
asbestos
fiber
over10
urn
in
length,
whichever-occurs
first.
b.
A
minimum
of
4
grid
openings
must
be
counted,
even
if
this
results
in
counting
more
than
100
asbestos
fibers
over
10
pm
in
length.
c.
The
grid
openings
examined
must
be
drawn
about
equally
from
a
minimum
of
3
specimen
grids.
9.
Counting
rules:
a.
Count
fibers
with
an
aspect
ratio
3:1.
b.
Count
a
fiber
bundle
as
a
single
fiber
with
a
width
equal
to
an
estimate
of
the
mean
bundle
width,
and
lengt
equal
to
the
maximum
length.
c.
Count
individual
asbestos
fibers
and
bundles
within
clusters
and
matrices,
as long
as
they
meet
the
definitions
of
fibers
and bundles
as
described
in 9A
and
9B.
d.
Count
the
fibers
which
intersect
the
top
andleft
sides
of
the
grid
opening
and
record
as
twice
their
visible
length.
Do
not
record
fibers
intersecting
the
bottom
and
right
sides
of
the
grid
opening.
e.
Count
only
one
end
of
the
fiber
to
avoid
possibly
counting
a
fiber
more
than
once.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
V
50
Recommended
Method
Modifications
10.
Fiber
identification
criteria:
a.
Each
fiber
suspected
to
be
chrysotile
must
first
be
examined
by
electron
diffraction
following
the
procedure
In
Figure
15
of
the
EPA
method.
If
the
characteristic
electron
diffraction
(ED)
pattern
is
observed,
the
fiber
shall
be
classified
as
CD
(chrysotile
identified
by
diffraction
pattern).
If
no
pattern
is
observed
or
the
pattern
is
not
distinctive,
the
fiber
shall
be
examined
by
EDXA
(energy
dispersive
x—ray
analysis)
and
classified
according
to
the
EPA
method.
Onily
chrysotile
fibers
classified
as
CD,
CMQ
(chrysotile
identified
by
morphology
and
semi—quantitative
EDXA)
or
CDQ
(chrysotile
identified
by
morphology,
electron
diffraction
and
semi—quantitative
EDXA)
shall
be
included
in
the
calculation
of
the
concentration
for
the
purposes
of
this
regulation.
b.
Each
fiber
suspected
to
be
amphibole
must
first
be
examined
by
electron
diffraction
following
the
procedure
in
Figure
18
of
the
EPA
Method.
Each
fiber
must
be
examined
by
EDXA.
-
If
a
random
orientation
electron
diffraction
pattern
showing
a
0.53
nm
layer
spacing
is
obtained,
and
the
elements
and
peak
areas
of
the
EDXA
spectrum
correspond
to
those
of
a
known
amphibole
asbestos,
the
fiber
shall
bç
classified
as
ADQ
(amphibole
identified
by
diffraction
and
semi—quantitative
EDXA).
If
the
random
orientation
electron
diffraction
pattern
cannot
be
obtained,
is
incomplete,
or
is
not
recognizable
as
a
non—
amphibole
pattern,
but
the
elements
and
the
peak
areas
of
the
EDXA
spectrum
correspond
to
those
of
a
known
amphibole
asbestos,
the
fiber
shallbe
classified
as
AQ
(amphibole
identified
by
semi—quantitative
EDXA).
Only
amphibole
fibers
classified
as
ADQ,
AQ,
AZQ
(amphibole
identified
by
zone
axis
electron
diffraction
and
semiquantitative
EDXA)
and
AZZQ
(amphibole
identified
by
2
zone
axes
electron
diffraction
and
semi—quantitative
EDXA)
shall
be
included
in,
the
calculation
of
asbestos
concentration.
11.
It
is
not
necessary
to
calculate
the
mass
concent’ation
of
asbestos
for
this
regulation.
Concentrations
must
be
reported
When
no
in
MFL>1O
pm.
asbestos
fibers
greater
than
10
pm
are
found,
report
MFL>1O
pm.
<0.2
•
Tech.
Notes
on
DW
Methods
October
1994
—
Section
V
51
Recommended
Method
Nodifications
METHOD
502.2
(R16),
USE
OF
THE
PID
This
Technical
Note
clarifies
when
a
photoionization
detector
(PID)
not
is
required.
Method
502.2,
Rev.
2.0
(R16).requires
the
use
of
a
PID
to
measure
volatile
organic
compounds
(VOCs)
that
cannot
be
measured
with
an
electrolytic
conductivity
detector.
If
only
halogenated
analytes,
such
as
the
trihalomethanes,
are
to
be
measured,
a
PID
is
not
needed.
This
option
will
al-low
laboratories
to
use
this
VOC method
for
determination
of
total
trihalomethanes
as
specified
at
fl41.30
without
the
expense
of
a
P10.
Tech.
Notes
on
Dli
Methods
-
October
1994
—
Section
V
52
Recommended
Method
Modifications
METHODS
502.2
(R16),
524.2
(R3) AND
551
(R15)
SAMPLE
DECHLORINATION
This Technical
Note
provides
guidance
to help
laboratories
correctly
dechlorinate
samples
for
compliance
with
the
total
trihaloniiethane
(TTHM)
monitoring
requirements
under
40
CFR
141.30
using
EPA
Method
502.2,
Rev.
2.0
(R16)
or 524.2,
Rev. 4.0
(R3)
or 551
(R15),
or
when
VOCs
and
THMs
are
to
be
measured
in
the same
sample.
This
guidance
also
applies
to
use
of
EPAMethods
502.1,
503.1
and
524.1
(R16).
These
methods
are
not
approved
for
THM
analysis
under
40
CFR
141.30,
but
some
laboratories
may
wish
to
use
these
methods
for
analysis
of
samples
other
than
compliance
samples.
This guidance
supersedes
the
discussion
on
ascorbic
acid
contained
in
the
introduction
(p.
3)
to
the
1991
EPA
manual
(R16).
The
Agency
believes
revised
guidance
is
warranted
because
laboratories
may
be
confused
bythe
variety
of
preservation
procedures
described
in
the
five
methods.
The
reagent
available
to
dechlorinate
samples
varies
with
the
method
used,
or
with
the
analyte
to
be
measured.
Laboratories
must
carefully
follow
the
preservation
procedure
described
in
each method,
especially
the
order
in
which
reagents
are
added
to
the
sample.
Each
method
allows
use
of
one
or
moredechlorination
reagents
depending
on
the
analyte
to
be
measured.
These
reagents
remain
available
for
use,
but
EPA
strongly
recommends
use
of
sodium
thiosulfate
as
the
dechlorination
reagent,
because
the
Agency
has
more
performance
data
demonstrating
the
effectiveness
of
this
chemical
than
for
other
dechlorination
reagents,.
One exception
•
vinyl
chloride
and
sodium
thiosulfate
the
analysis.
EPA
be
acidified
iinmedi
as
for analysis
of
551
(R15).
to
this
recommendation
is
ascorbic
acid
must
be
used
when
Qther
gases
are
measured
with
a
mass
spectrometer,
because
in
an
acidified
sample
generates
a
gas
that
interferes
with
cautions
that
samples
dechlorinated
with
ascorbic
acid
must
ately,
as
directed
in
the
method.
Other
exceptions,
such
haloacetonitriles
are
described
in
Section
8
of
EPA
Method
Tech.
Iotes
on
Dl’!
Methods
October
1994
—
Section
V
53
Recommended
Method
Modifications
METHOD
504.1
(R5),
CHROMATOGRAPHIC
INTERFERENCES
Although
this
Technical
Note
discusses
misidentifications
that
may
occur
when
measuring
1,2—dibromoethane
(EDB)
or
dibromochioropropane
(DBCP)
with
Method
504.1
(R5),
the
guidance
and
warnings
provided
here
are
applicable
to
the
interpretation
of
analytical
results
from
any
method.
Volatile
organic
chemicals
(VOCs)
or
trihalomethanes
(THt1s)
can
cause
chromatographic
interference
problems
if
these
chemicals
•are
in
the
sample,
and
coelute
on
the
column
used
to
separate
and
identify
EDB
or
DBCP.
Interferences
can
lead
to
false
positive
results,
if
a
coeluting
VOC
or
THM
is
misidentified
as
EDB
DBCP.
or
Since
any
method,
even
one
that
uses
a
selective
detector,
is
subject
to
false
positive
results,
any
result
that
exceeds
an
actionconcentration
must
be
rigorously
Method
504.1
uses
an
electron
capture
detector
that
is
very.
sensitive
and
stable.
Although
this
detector
is
exdellent
at
detecting
very
low
cbncentrations
of
halogenated
compounds,
it
is
subject
to
many
interferences.
Sections
4.3
and
6.6.2
in
Method
504.1
note
that
a
common
THM
disin
fection
by—product
in
chlorinated
water
supplies,
dibromoc:hloromethane,
can
elute
close
to
EDB.
This
means
in
the
initial
demonstration
of
capability,
a
laboratory
must
determine
the
retention
time
of
dibromochloromethäne
or
other
compounds
that
might
coelute
with
the
method
analytes.
A
relative
response
factor
and
retention
time
for
each
possible
interfering
anialyte
should
be
determined.
These
retention
times
can
be
determined
by
using
procedures
in
,
Method
551
to
prepare
and
analyze
THM
and
VOC
standards
for
analysis
on
a
Method
504.1
chromatographic
column.
This
information
can
be
obtained
more
easily
if
a
DB—1
column
is
used
in
Method
504.1
and
the
retention
times
are
compared
to
the
THM
and
VOC
retention
times
obtained
with
the
DB—1
column
used
in
Method
551.
Confirmation
procedures
must
be
followed
before
taking
action
on
a
result.
Confirmation
of
potential
Method
504.1
or
Method
551
chromatographic
interferences
can
be
obtained
with
an
inexpensive
purge—and—trap
analysis
(EPA
Method
502.2
(R16)
or
524.2
(R3)).
These
methods
can
identify
interfering
trihalomethanes,
or
VOCs
that
might
occur
with
EDB
if
the
‘source
of
EDB
were
unleaded
gasoline
(cf.
Sect.
2.3).
Although
Method
524.2
‘is
not
as
sensitive
as
Method
504.1,
EDB
can
be
measured
at
concentrations
greater
than
0.06
pg/L.
Other
confirmation
procedures,
which
are
described
in
Method
504.1,
are:
analysis
on
a
second
column
with
dissimilar
retention
times
(Sect.
6.6.2),
and
changing
the
temperature
program
to
provide
sufficient
separation
between
EDB
and
dibromochioromethane
(Sect.
9.1.2).
EPA
emphasizes
that
knowledge
of
probable
contaminants
‘in
a
sample,
and
of
method
interferences
are
key
parts
of
quality
assurance
and
good
data
interpretation
when
using
ji
analytical
method.
Laboratories
reporting
data
must
realize
that
interpreters
of
occurrence
data
are
often
unfamiliar
with
weaknesses
in
an
analytical
method,
and
that
officials
may
enforce
on
the
data
as
provided
by
the
laboratory.
EPA
strongly
encourages
data
reviewers
to
Tech.
Notes
on
DW
Methods
October
1994
—
Section.V
54
Recommended
Method
Modifications
question
the
plausibility,
not
just
the
possibility,
of
a:
result,
and
not
assume
that
a
laboratory
has
always
eliminated
analytical
error.
A
skeptical
approach
is
especially
important
when
initial
sample
results
are
being
interpreted.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
V
55
Recommended
Method
Modifications
METHODS
505,
507,
508
(R16),
INTERCHANGE
OF
DETECTORS
This
Technical
Note
clarifies
under
what
conditions
a
laboratory
may
use
either
an
electron—capture
detector
(ECD)
or
a
nitrogen—phosphorous
detector
(NPD)
in
EPA
Methods
505,
Rev.
2.0;
507,
Rev.
2.0;
or
508,
Rev.
3.0
(R16).
Laboratories
may
wish
to
use
a
different
detector
to
decrease
method
detection
limits.
For
example,
use
of
an
NPD
in
Method
505
can
increase
the
sensitivity
of
the
analysis
for
alachior,
atrazine
and
simazine.
Section
6.8.3
of
Methods
507
and
508
and
Sect.
10.4
of
Methods
505,
507
and
508
allow
use
of
an
LCD
NPD
or
detector
provided
the
initial
demonstration
of
capability
criteria
are
met.
These
criteria
are
specified
in
Section
10
of
each
method.
Section
6.8.3
of
Methods
507
and
508
note
that
a
mass
spectrometer
might
be
used.
This
Note
withdraws
this
recommendation,
which
was
made
before
Method
525.2
was
available.
EPA
no
longer
recommends
use
of
a
mass
spectrometer
with
Methods
507
and
508,
because
important
tuning
and
calibration
procedures
for
the
mass
spectrometer
are
not
described
in
either
method,
and
because
Method
525.2
thoroughly
describes
these
procedures.
Method
525.2
is
approved
for
determination
of
all
Method
507
and
508
analytes,
except
PCBs
as
the
seven
Aroclors.
Tech.
Notes
on
OW
Methods
October
1994
—
Section
V
56
Recommended
Method
Modifications
EPA
METHODS
507,
508,
515.1
(R16),
MERCURIC
CHLORIDE
This
Technical
Note
removes
the
requirement
to
use
mercuric
chloride,
because
concerns
have
been
raised
about
the
environmental
hazards.
and
costs
associated
with
disposal
of
mercuric
compounds.
Mercuric
chloride
is
used
as
a
biocide
in EPA
Methods
507,
Rev.
2.0;
508,
Rev.
3.0;
and
515.1,
Rev.
4.0
(R16).
Since
drinking
water
usually
exhibits
limited
biological
activity,
EPA
is
removing
the
requirement
under
Sect.
8.2
of
Methods
507,
508,
and
515.1
to
use
mercuric
chloride
as
a
bactericide.
To
minimize
the
possibility
of
occasional
false—negative
results,
the
Agency
would
still
require
the
use
of
mercuric
chloride
in
any
drinking
water
sample
that
might
be
expected
to
exhibit
biolog.ical
degradation
of
a
target
pesticide.
There
are
also
environmental
and
economic
concerns
about
addition,
of
acid
to
drinking
water
samples
in
the
VOC
methods
(Methods
502.2,
524.2,
arid
551).
However,
EPA
will
not
remove
this
requirement,
because
EPA
has
data
that
demonstrates
microbiological
-degradation
of
VOCs
in
drinking
water
samples.
Tech.
Notes
on
DW
Methods
October
1994
—
Section
V
57
Recommended
Method
Modifications
EPA METHOD
1613,
DIOXIN
(R17)
This Technical
Note does
not
change
Method
1613
(R17).
It
describes
how
to
make some steps
in
the
method
specifically
applicable
to
measurement
of
2,3,7,8—tetrachlorodibenzo—p—dioxin
(TCDD).
Guidance
is
needed
because
Method
1613
was
written
to
determine
many
isomers
of
dioxins
and
furans,
but
under
the
Safe
Drinking
Water Act,
EPA
only
regulates
the
2,3,7,8—TCDD
isomer.
Also,
information
to
determine
if
the
drinking
water
sample
needs
to
be
filtered
is
not
clearly
provided
in
Method
1613.
Using
this
guidance
will
substantially
decrease
the cost
of
Method
1613,
because
it
eliminates
many
costly
steps that
are
not required
when
only
TCDD
is
to
be
determined.
EPA
METHOD
1613
OGWDW
GUIDANCE
AND
CLARIFICATION
FOR
ANALYSIS
OF
2,3,7,8—TETRACHLORODIBENZO—p—DIOXIN
(TCDD)
IN
DRINKING
WATER
1.
The
only isotopically
labeled
compounds
which
are
necessary
for
calibration
and
quantitation
in
addition
to
the
native
2,3,7,8—TCDD
are
13
C
12
2,3,7,8—TCDD
the spiking
compound),
37
C1
4
2,3,7,8—TCDD
(the
clean
up
standard),
and
3
C,
2
1,2,3,4—TCDD
(the
internal
standard).
2.
During
calibration,
selected
ion
current
profiles
of
only the
compounds
in
item
1
above
need
be
obtained
according
to
directions
in
Sect.
7
of
the method
by
monitoring
the
exact masses
specified
for
these
compounds
in
Table
3
of
the
method
at
>10,000
resolving
power..
The
relative
abundances
must met
the
criteria
specified
in
the
method.
There
must
be
at
least
baseline
resolution
in
the
chromatogram
between
the
1,2,3,4—
TCDD
and
the 2,3,7,8—TCDD
isomers.
3.
If
the sample
is
colorless,
odorless,
has
a
turbidity
of
one
(1)
NTU
or
.less
and
consists
of
a
single
phase,
filtration
is
not
required,
and
the
sample
may be
analyzed
according
to
Sect.
11.1
of
the
method.
Turbidity
must be
measured
with
an
approved
method.
Any
sample
containing
multiple
phases,
or
having
a
turbidity
.of
more
than
one
(>1)
NTU
must
be
filtered.
The
filter
particulate
must
be
analyzed
according
to
Sect.
11.2
of
the
method.
4.
Since drinking
water
samples
are
relatively
free
frOm
interferences,
the
optional
clean—up
steps
described
in
the
method
probably
will
not
be
needed
for
most samples.
Tech.
Notes
on
DW
Methods
October
1994
—.
Section
V
58
Recommended
. Method
Modifications
SECTION
VI.
EPA
CONTACTS
AND
METHOD
REFERENCES
OBTAINING
METHODS AND
TECHNICAL
ASSISTANCE
For
assistance
in
obtaining
copies
of
EPA
methods,
‘Dr
for
answers
to
technical questions
about
drinking
water
methods
please
contact:
U.S.
EPA,
Environmental Monitoring
Systems
Laboratory
Chemistry
Research
Division
(MC
564)
Cincinnati,
OH
45268—0001
Telephone:
513
569—7586
CERTIFICATION
AND
REGULATORY
ASSISTANCE
For
answers
to
questions
about
laboratory
certificahion,
the
Labcert
Bulletin,
and
the
regulatory status
of
drinking
water
methods
please
contact:
U.S.
EPA,
Technical Support
Division
Drinking
Water
Quality Assessment
Branch
(MC
140)
ATTN:
Methods
and
Laboratory
Certification
Cincinnati, OH
45268—0001
Telephone:
513
569—7938
REFERENCES
Ri.
Approved
EPA
Methods
200.7,
200.8,
200.9,
and
245.1
are
contained
in
“Methods
for
the
Determination
of
Metals
in
Environmental
Samples
—
Supplement
I,”
May
1994,
NTIS
PB94—184942.
R2.
EPA
Method
100.2,
“Determination
of
Asbestos
Structures
over
10pm
in
Length
in
Drinking
Water,”
June
1994,
NTIS
PB94—201902.
R3.
Approved
EPA
Methods
515.2,
524.2,
548.1,
549.1,
552,1
and
555
are
contained
in
“Methods
for
the
Determination
of
Organic
Compounds
in
Drinking Water
— Supplement
II,”
August 1992,
NTIS
PB92—207703.
R4.
Approved
EPA
Methods 180.1,
300.0,
335.4,
353.2
and
‘ecommended
Method
375.2
are
contained
in
“Methods
for
the Determination
of
Inorganic
Substances
in
Environmental
Samples,”
August
1993,
NTIS
PB94—121811.
R5.
EPA
Method
504.1,
“1,2—Dibromoetháne
(EDB),
1,2—Dibromo--3—chloropropane
(DBCP),
and
1,2,3—Trichloropropane
(123TCP)
in
Water
by
Microextraction
and
Gas
Chromatography,”
1993.
R6.
EPA
Method
508.1, Rev.
1.0,
“Determination
of
Chlorinated
Pesticides,
Herbicides, nd
Organohalides
by
Liquid—Solid
Extraction
ad
Electron
Capture
Gas
Chromatography,”
1994.
R7.
EPA
Method
525.2,
Rev.
1.0,
“Determination
of
Organlé
Compounds
in
Drinking
Water
by
Liquid—Solid
Extraction
and
Capillary
Column
Gas
Chromatography/Mass Spectrometry,”
March
1994.
59
R8.
Method
6610 “Carbamate
Pesticides”
is
contained
in
Standard
Methods
for
the
Examination
of
Water
and
Wastewater
18th
Edition
Supplement,
1994
may
be
purchased
from
the
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
R9.
GLI
Method
2,
“Turbidity”
is
available
free
from
Great
Lakes
Instruments,
Inc.,
November
2,
1992.
Rio.
Orion
Technical
Bulletin
601
“Standard
Method
of
Test
for
Nitrate
in
Drinking
Water,”
July
1994
is
identical
to
Orion
WeWWG/5880,
which
had
previously
been
approved
for
nitrate
analysis
at
40
CFR
141.23(k)(1).
ATI
Orion
republished
this
method
in
1994,
and
renumbered
it
as
601,
because
the
1985
manual
“Orion
Guide
to
Water
and
Wastewater
Analysis:,”
which contained
WeWWG/5880,
is
no
longer
available.
Technical
Bulletin
601
is
available
free
from
ATI
Orion,
529
Main
Street,
Boston,
MA
02129.
Laboratories
wishing
to
use
the
Orion
method
should
be
aware
that
SM
4500—NO
3
—D,
which
is
published
in
the
18th
edition
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
is
equivalent
to
Orion
60.1.
Ru.
The
American
Society
for
Testing
and
Materials
(ASTM)
annually
reprints
all
of
the
methods
contained
in
the
Annual
Book
of
ASTM
Methods,
Vols.
11.01
and
11.02,
including
methods
that
have
not
been
editorially
or
technically
revised.
Thus,
it
is
permissible
to
use
any
edition
that
contains
the
EPA—approved
version
of
the
method
that
is
approved.
The
Annual Book
of
ASTM
Methods
may
be
purchased
from
ASTM,
1916
Race
Street,
Philadelphia,
PA
19103.
R12.
Eighteenth
edition
of
Standard
Methods
for
the
Examination
of
Water
and
Wastewater,
1992
may
be
purchased
from
the
American
Public
Health
Association,
1015
Fifteenth
Street
NW,
Washington,
D.C.
20005.
R13.
EPA
Method
245.2,
“Mercury,
Automated
Cold
Vapor
Technique,”
Environmental
Monitoring
Systems
Laboratory,
Cincinnati,
OH
45268,
1974.
Also
contained
in
reference
14.
R14.
“Methods
for
Chemical
Analysis
of
Water
and
Wastes,”
EPA,
March
1983,
NTIS PB84—128677.
R15.
Approved
EPA
Methods
506,
547,
550,
550.1
and
551
are
contained
in
“Methods
for
the
Determination
of
Organic
Compounds,
in
Drinking
Water
——
Supplement
I,”
July
1990,
NTIS
PB91-146027.
R16.
Approved
EPA
Methods
502.2, 505,
507,
508,
508A,
515.1
and
531.1,
and
Methods
502.1,
503.1,
and
524.1,
which
will
be
withdrawn
are
contained
in
“Methods
for
the
Determination
of
Organic
Compounds
in
Drinking
Water”
December
1988,
Revised
July
1991,
NTIS
PB91—231480.
R17.
EPA
Method
1613,
Revision
B,
“Tetra—through—Octa—
Chlorinated
Dioxins
and
Furans
by
Isotope—Dilution
HRGC/HRMS,”
October
1994,
NTIS
PB95—104774.
60
R18.
EPA Method 100.1,
“Analytical
Method
for
the
Determination
of
Asbestos
Fibers
in
Water,”
September
1983,
NTIS
PB83—260471.
R19.
Methods
1—3300—85,
1—1030—85,
1—1601—85,
1—2598—85,
1—1700—85
and
1—2700—
85
in
Techniques
of
Water
Resources
Investigations
of
the
U.S.
Geological
Survey,
Book
5,
Chapter
A—i,
3rd
ed.,
U.S.
Geological
Survey,
Books
and
Open
File Reports
Section,
Box
25425,
Federal
Center,
Denver,
CO
80225—
0425, 1989.
R20.
“Waters
Test Method
for
Determination
of
Nitrite/Nitrate
in
Water
Using
Single
Column
Ion Chromatography,”
Method
8—1011
is
available
free
from
Millipore
Corporation,
Waters
Chromatography
Division,
34
Maple
Street,
Milford,
MA
01757.
R21.
Industrial
Method
No.
129—71W,
“Fluoride
in
Water
arid
Wastewater,”
December
1972, and
Method
No.
380—75WE,
“Fluoride
in
Water
and
Wastewater,”
February
1976
are
available
free
from
Technicon
Industrial
Systems,
Tarrytown,
NY 10591.
V
R22.
Method
1—2601—90
in
Methods
of
Analysis
by
the
U.S.
Geological
Survey
National
Water
Quality
Laboratory——Determination
of
Inorganic
and
Organic
Constituents
in
Water
and
Fluvial
Sediments,
Open
File
Report
93—125
is
available
from
U.S.
Geological
Survey,
Books
and
Open
File
Reports
Section,
Box 25425,
Federal
Center,
Denver,
CO
80225—0425,
1993.
References
Ri
to
R4
are
available
for
a fee
through
:the
National
Technical
Information
Service
(NTIS),
which
is
located
at
U.S.
Department
of
Commerce,
5285
Port
Royal
Road,
Springfield,
Virginia
22161;
the
toll—free
number
is
(800)—553—6847.
Until
references
R5
to
Ri
are
published
in
“Methods
for
the
Determination
of
Organic
Compounds
in
Drinking
Water
—
Supplement
III,”
these
methods
are available
free
from
EPA—EMSL—Cincinnati,
Cincinnati,
OH
45268.
The
phone
number
is
(513)
569—7586.
The
“Supplement
III”
manual
is
expected
to
be
published
by
EMSL—Cincinnati
in
late
1995.
61
a.$,
GOVVNNT
PRINTING
OPPICE;
1995—65O—OOj/OO22o
/41i.;L.
/11/
This
ASTM D6508,
Rev2 method
document
has
been
reviewed by EPA Office
of Drinking
Water
and
Wastewater for EPA
Tier
3
approval. Added updated
QC
criteria based
upon
statistical
analysis
by Dyncorp.
ATP Case
#:
N00-0002
and D00-0002
Draft
#:
Second
draft with
EPA
Modifications: ASTM D6508, Rev 2
Date:
December 2000
Method Author:
Jim
Krol
Telephone:
508/482-2131
FAX:
508/482-3625
Test
Method
for
Determination
of
Dissolved
Inorganic
Anions
in Aqueous
Matrices
Using
Capillary Ion
Electrophoresis
and Chromate
Electrolyte
V
1 Scope
1.1 This
test method covers the determination
of
the
inorganic anions fluoride,
bromide,
chloride, nitrite, nitrate,
ortho-phosphate, and sulfate in drinking
water,
wastewater, and
other aqueous
matrices using capillary
ion electrophoresis
(CIE) with
indirect UV detection.
See Fig. 1 through
6.
1.2 The
test method uses
a chromate-based electrolyte
and indirect UV detection
at
254
nm. It is
applicable
for the determination
of inorganic anions
in the
range of 0.2 to 50 mgJL except for fluoride
whose range is 0.2 to 25
mgJL.
1.3 It is the
responsibility
of
the
user to ensure the
validity of this test
method for
other anion concentrations and untested
aqueous matrices.
Note 1: The
highest accepted anion
concentration submitted for
P&B extend the anion
concentration range for the following
anions; Chloride to
93 mg/L, Sulfate to 90
mgJL,
Nitrate to 72 mg/L, and ortho-phosphate to
58
mg/L.
1.4 This
method does not purport
to address all of
the safety
problems,
if
any,
associated with its use, It is the
responsibility
of
the user
of this standard
to
establish
appropriate
safety
and health practices
and determine the
applicability
of
reciulatorv
limitations
prior
to use.
For specific hazard
statements,
see
sec.
9.
2
2 Referenced
Documents
2.1 ASTM
Standards
D
1066
Practice
for
Sampling
Steam1
1
D
1129
Terminology
Relating
to Water
D 1193
Specification
for Reagent
Water
1
D 2777
Practice
for Determination
of
Precision
and Bias
of
Applicable
Methods
of
Committee
D-1
9
on Water
1
D
3370
Practices
for Sampling
Water’
D
3856
Guide for
Good
LaboratorX
Practices
in Laboratories
Engaged
in
Sampling
and
Analysis of
Water
D
5810
Standard
Practice
of
Spiking
Samples’
D
5847
Standard
Practice for
Writing
Quality
Control
Specifications
for
Standard
Test
Methods
for
Water Analysis
1
D
5905
Standard
Specification
for Substitute
Wastewater
1
F 488
Test
Method
for Total
Bacterial
Count in
Water
2
2.2
EPA 40 CFR
Ch.1
(7-1-92 Edition),
Pt
136, App.
B, page
565
—567:
Definition
and Procedure
for the
Determination
of the
Method
Detection
Limit-Revision
1.11.
2.3
Draft Protocol
for EPA
Approval
of New Methods
for
Organic
and Inorganic
Analytes
in
Wastewater
and
Drinking
Water,
dated
Mar
1999, EPA-821-B-98-003.
3
Terminology
3.1
Definitions
-
For definitions
of terms
used in
this test method,
refer
to
Terminology
D1129.
3.2
Description
of
Terms Specific
to
This
Test Standard:
3.2.1 Capillary
Ion Electrophoresis
-- an
electrophoretic
technique
in which
an
UV
absorbing
electrolyte
is placed
in a
50
pm
to 75
pm fused
silica capillary.
Voltage
is
applied across
the
capillary
causing
electrolyte
and anions
to
migrate
towards
the anode
and
through
the
capillary’s
UV detector
window.
Anions
are separated
based
upon
the
their
differential
rates
of migration
in
the
electrical
field.
Anion detection
and quantitation
are
based
upon the
principles
of
indirect
UV
detection.
3.2.2 Electrolyte
-- combination
of a
UV absorbing
salt
and
an electroosmotic
flow
modifier
placed
inside the
capillary,
used
as a
carrier
for
the analytes,
and
for
detection
and
quantitation.
The
UV
absorbing
portion
of
the
salt must
be
anionic
and
have
an electrophoretic
mobility
similar
to
the analyte
anions
of interest.
3.2.3
Electroosmotic
Flow (EOF)
--
the
direction
and
velocity
of
electrolyte
solution
flow
within
the capillary
under
an applied
electrical
potential
(voltage);
the
velocity
and
direction
of flow
is determined
by electrolyte
chemistry,
capillary
wall
chemistry,
and
applied
voltage.
3.2.4
Electroosmotic
Flow
Modifier
(OFM)
-- a cationic
quaternary
amine
in the
electrolyte
that dynamically
coats the
negatively
charged
silica
wall giving
it
a
net
positive
charge.
This reverses
the
direction
of the
electrolyte’s
natural
electroosmotic
flow
and
directs
it towards
the anode
and
detector.
This modifier
augments
anion
migration
and
enhances
speed of
analysis.
Its
concentration
secondarily
effects
anion
selectivity
and
resolution.
See
Fig. 7.
1) Annual
Book of
ASTM
Standards,
Vol. 11.01
2) Annual
Book of
ASTM
Standards,
Vol. 11.02
3
3.2.5
Electrophoretic
Mobility -- the specific velocity of
a charged analyte in
the
electrolyte
under specific electroosmotic flow conditions. The mobility of
an
analyte
is
directly related to
the
analyte’s equivalent ionic conductance
and
applied voltage, and is the primary mechanism of separation.
3.2.6
Electropherogram
-- a
graphical
presentation of UV
detector
response
versus
time
of
analysis;
the x
axis
is
migration time which is used
to qualitatively
identify
the anion,
and the
y
axis is
UV
response which
can be
converted
to
time
corrected peak
area
for
quantitation.
3.2.7
Hydrostatic Sampling
-- a
sample introduction
technique
in
which the capillary
with electrolyte
is
immersed
in the
sample, and both
are
elevated
to
a specific
height, typically
10 cm, above
the
receiving
electrolyte
reservoir
for a preset
amount of
time, typically less than
60
s.
Nanolitres
of
sample
are
siphoned into
the
capillary by
differential
head
pressure
and gravity.
3.2.8 Indirect UV
Detection -- a
form
of
UV detection in
which the
analyte displaces
an
equivalent
net charge
amount of
the
highly UV absorbing component
of
the
electrolyte
causing
a net
decrease
in
background absorbance. The magnitude
of the decreased
absorbance is
directly proportional
to analyte concentration.
Detector output
polarity is reversed in order
to
obtain a
positive mV response.
3.2.9.Midpoint of
Peak Width
-- CIE
peaks
are
typically asymmetrical
with the
peak
apex
shifting
with
increasing concentration,
and peak
apex
may not
be
indicative of
true
analyte migration time.
Midpoint of
peak
width
is the
midpoint
between the analyte peak’s
start
and
stop
integration,
or
the peak center
of
gravity.
3.2.10
Migration
Time
--the time required
for a
specific
analyte
to
migrate
through
the
capillary to
the detector. The
migration time in capillary
ion
electrophoresis is
analogous
to retention
time
in
chromatography.
3.2.11 Time Corrected
Peak Area
-- normalized peak area;
peak area divided
by
migration
time.
CE
principles state
that peak
area is dependent
upon
migration
time, i.e.
for
the
same concentration
of analyte, as
migration time
increases
(decreases) peak area increases (decreases).
Time corrected
peak
area
accounts for
these changes.
4
Summary
of
Test Method
4.1 Capillary
ion
electrophoresis, see
Fig. 7
through Fig. 10,
is a
free zone
electrophoretic technique optimized
for the
determination
of
anions
with
molecular
weight
less than 200. The anions
migrate and
are separated according
to their
mobility
in
the electrolyte when
an electrical field is
applied through
the
open
tubular fused
silica
capillary. The
electrolyte’s
electroosmotic low
modifier
dynamically coats the inner wall of the
capillary
changing the surface
to
a net
positive charge. This reversal of
wall
charge
reverses
the
natural
EOF.
The
modified
EOF in combination with
a negative
power supply augments
the
mobility
of the analyte
anions towards the
anode
and detector achieving
rapid
analysis
times. Cations migrate in the opposite
direction
towards the cathode
and
are
removed from the sample during
analysis. Water
and other neutral
species
move
toward the
detector at
the same rate
as the EOF.
The neutral species
migrate
slower than the analyte anions and do
not interfere
with anion analysis.
See Fig.
7 and
8.
4
4.2 Sample
is
introduced into the
capillary
using
hydrostatic
sampling.
The
inlet
of
the
capillary
containing
electrolyte
is immersed
in
the
sample
and
the
height
of the
sample
raised
10
cm for
30
s where
low
nanolitre
volumes
are
siphoned
into
the
capillary.
After
sample
loading,
the
capillary
is immediately
immersed
back
into
the
electrolyte.
The
voltage
is applied
initiating
the
separation
process.
4.3
Anion
detection
is based
upon the
principles
of indirect
UV detection.
The
UV
absorbing
electrolyte
anion
is
displaced
charge-for-charge
by the
separated
analyte
anion.
The
analyte
anion
zone
has a
net decrease
in background
absorbance.
This
decrease
in UV
absorbance
is
quantitatively
proportional
to
analyte
anion
concentration.
See
Fig.
9.
Detector
output
polarity
is
reversed
to
provide
positive
mV
response
to the
data system,
and to
make the
negative
absorbance
peaks
appear
positive.
4.4
The
analysis
is
complete
once
the
last
anion
of
interest
is detected.
The
capillary
is automatically
vacuum
purged
by the
system
of
any
remaining
sample,
and
replenished with fresh
electrolyte.
The
system
is now
ready
for
the
next
analysis.
5
Significance
and
Use
5.1
Capillary
ion electrophoresis provides
a
simultaneous
separation
and
determination
of
several
inorganic
anions
using
nanolitres
of
sample
in
a
single
injection.
All
anions
present
in
the sample
matrix
will
be
visualized
yielding
an
anionic
profile
of the
sample.
5.2 Analysis
time is
less
than
5 mm
with
sufficient
sensitivity
for
drinking
water,
and
wastewater
applications.
Time
between
samplings
is less
than
7 minutes
allowing
for
high
sample
throughput.
5.3
Minimal
sample
preparation
is
necessary
for
drinking
water
and
wastewater
matrices.
Typically
only a
dilution
with
water is
needed.
5.4 This
test
method
is
intended
as
an alternative
to other
multi-analyte
methods
and
various
wet chemistries
for the
determination
of inorganic
anions
in
water
and
wastewater.
Compared
to
other multi-analyte
methods
the
major
benefits
of
CIE
are
speed
of
analysis,
simplicity,
and reduced
reagent
consumption
and
operating
costs.
6
Interferences
6.1
Analyte
identification,
quantitation,
and
possible
comigration
occur
when
one
anion
is
in
significant
excess
to
other
anions
in
the sample
matrix.
For
two
adjacent
peaks,
reliable
quantitation
can
be achieved
when
the concentration
differential
is less
than 100:1.
As
the
resolution
between
two
anion
peaks
increase
so does
the
tolerated
concentration
differential.
In samples
containing
1000
mgIL Cl,
1 mgJL
SO
4
can be
resolved
and
quantitated,
however,
the high
Cl
will
interfere
with
Br
and
NO
2quantitation.
6.2 Dissolved
carbonate,
detected
as
HC0
31
,
is
an anion
present
in
all aqueous
samples,
especially
alkaline
samples.
Carbonate
concentrations
greater
than
500
mg/L
will interfere
with
PC
4
quantitation.
6.3
Monovalent organic
acids,
except
for
formate,
and
neutral
organics
commonly
found
in wastewater
migrate
later
in the electropherogram,
after
carbonate,
and
do not
interfere.
Formate,
a
common
organic
acid
found
in environmental
samples,
migrates
shortly
after
fluoride
but
before
phosphate.
Formate
concentrations greater
than
5 mg/L
will interfere
with
fluoride
identification
and
quantitation.
Inclusion
of
2 mgIL
formate
into
the
Mixed
Anion
Working
Solution
aids
in
fluoride
and
formate
identification
and quantitation.
5
6.4
Divalent organic
acids usually
found
in wastewater
migrate
after
phosphate.
At
high
concentrations,
greater
than
10
mgJL,
they
may
interfere
with
phosphate
identification
and quantitation.
6.5
Chlorate
also
migrates
after phosphate
and
at concentrations
greater than
10
mgJL
will
interfere
with phosphate
identification
and
quantitation.
Inclusion
of
5
mglL chlorate
into the
Mixed Anion
Working
Solution
aids in phosphate
and
chlorate
identification
and
quantitation.
6.6
As
analyte
concentration
increases,
analyte
peak
shape
becomes
asymmetrical.
If
adjacent
analyte
peaks are
not baseline
resolved,
the data
system
will drop
a
perpendicular
between
them
to
the
baseline.
This causes
a
decrease
in
peak
area
for
both
analyte peaks
and
a
low bias
for
analyte
amounts.
For optimal
quantitation,
insure that
adjacent
peaks
are fully
resolved,
if they
are
not,
dilute
the sample
1:1 with
water.
6.7
Samples
containing
high
levels
of
TOC, total
organic
carbon,
may
effect
the
observed
analyte migration
times.
The
TOC
binds
to
the
capillary
surface
decreasing
the
EOF
and increasing
analyte
migration
times. Refer
to Figure
7.
However,
the change
in EOF
does not
effect
analyte
selectivity.
Analytes
are
identified
using
normalized
analyte
migration
times
with respect
to
a reference
peak,
chloride,
always
the
first
peak
in the
electropherogram.
The
surface
can be
regenerated
with
a 5
minute wash
with
500 mM
NaOH.
7
.Apparatus
7.1
Capillary
Ion Electrophoresis
System
-- the
system
consists
of
the following
components,
as shown
in Fig. 10,
or
equivalent:
3
7.1.1
High
Voltage
Power
Supply
-- capable
of generating
voltage
(potential)
between
0
and
minus 30
kV
relative
to ground
with the
capability
working
in
a
constant
current
mode.
7.1.2
Covered
Sample Carousel
--
to prevent
environmental
contamination
of
the
samples
and electrolytes
during
a multi-sample
batch
analysis.
7.1.3
Sample
Introduction
Mechanism
-- capable
of
hydrostatic
sampling
technique,
using
gravity,
positive
pressure,
or
equivalent.
7.1.4
Capillary
Purge
Mechanism
-- to
purge the
capillary
after every
analysis
with
fresh
electrolyte
to eliminate
any
interference
from
the
previous
sample
matrix,
and to
clean
the
capillary
with
other
reagents,
such
as
sodium
hydroxide.
7.1.5 UV Detector
--
having
the
capability
of monitoring
254 nm,
or equivalent,
with
a
time
constant
of
0.3
S.
7.1.6 Fused
Silica
Capillary
--
a 75
pm
(inner
diameter)
x 375 pm
(outer
diameter)
x
60 cm
(length)
having
a polymer
coating
for flexibity,
and
a non-coated
section
to act as
the cell window
for UV
detection.
7.1.7
Constant
Temperature
Compartment
-- to
keep the
samples,
capillary,
and
electrolytes
at
constant
temperature.
3)
Available
from Waters,
34 Maple
St., Milford,
Ma.,
01757,
800/252-4752.
6
7.2
Data
System
-- computer
system
that can
acquire data
at 20
points
per second
minimum,
express
migration
time
in
minutes
to
3
decimal
places,
use
midpoint
of
the
analyte
peak
width,
or center
of
gravity,
to determine
the
analyte
migration
time,
use normalized
migration
times
with respect
to a
reference
peak for
qualitative
identification,
use
time corrected
peak
area
response
for analyte
quantitation,
and
express
results in
concentration
units.3
Note 2:
It is
recommended
that integrators
or
standard
chromatographic
data processing
not
be
used with
this
test
method.
7.3
Anion
Exchange
Cartridges
in
the
Hydroxide
form.4
7.4
Plastic
Syringe
-- 20
mL, Disposable.
7.5 Vacuum
Filtration
Apparatus
-- capable
for filtering
100
mL of
reagent
through
a
0.45 m
aqueous
filter.
8
Reagents
and
Materials
8.1
Purity
of
Reagents:
-- Unless
otherwise
indicated,
it is
intended
that all reagents
shall conform
to the reagent
grade
specification
of
the
Analytical
Reagents
of the
American
Chemical
Society,
where
such specifications
are
available.
Other
grades
may be
used,
provided
it
is first
ascertained
that
the
reagent
is
of
sufficient
high
purity
to permit
its use without
lessening
the
performance
or accuracy
of the
determination.
Reagent chemicals
shall
be used
for all tests.
Note
3:
Calibration
and detection
limits
of
this
method
are
biased
by
the
purity
of
the
reagents.
8.2
Purity
of
Water:--
Unless
otherwise
indicated,
references
to water
shall
be
understood
to mean
Type I reagent
water
conforming
or
exceeding
specification
Dli 93.
Freshly
drawn
water
should
be
used
fr preparation
of
all stock
and
working
standards,
electrolytes,
and
solutions.
Performance
and
detection
limits
of this
method
are
limited by
the purity
of
reagent
water, especially
TOC.
8.3
Reagent Blank:
Reagent
Water
or any other
solution
used to
preserve
or
dilute
the sample.
8.4 Individual
Anion
Solution,
Stock:
Note
4: It is
suggested
that certified
individual
1000 mg/L
anion
standards
be
purchased
for use
with this
test method.
Note
5: All
weights
given
are for anhydrous
or
dried salts.
Must account
for reagent
purity to
calculate
true value concentration.
Certify
against
NIST
traceable
standards.
8.4.1
Bromide
Solution,
Standard
(1.0
mL = 1.00
mci
Bromide):
Dry
approximately
0.5
g
of
sodium bromide
(NaBr) for
6
h at 150°C
and
cool
in
a
desiccator.
Dissolve
0.128
g
of the dry
salt in
a 100
mL
volumetric
flask
with
water,
and
fill to
mark with
water.
4)
Available
from
Alltech
Associates,
PIN 30254,
2051 Waukegan
Rd,
Deerfield
IL., 60015,
847/948-
8600.
5)
Reagent
Chemicals,
American Chemical
Society
Specifications,
Am. Chem.
Soc.,
Washington,
DC
For suggestions
on
the testing
of reagents
not listed
by the American
Chemical
Society,
see
Analar
Standards
for
Laboratory
Chemicals,
BDH
Ltd.,
Poole,
Dorset.
U.K., and
the
United
States
Pharmacopeia
and
National
Formulary,
U.S.
Pharmacopoeia
Convention,
Inc. (USPC),
Rockville,
Md.
6)
Although
the
reagent
water may
exceed
Dli 93 specification,
the
reagent water
needs to
be
periodically
tested for
bacterial
contamination.
Bacteria
and their
waste products
may
adversely
affect
system
performance.
As
a
guide,
ASTM
type
IA water
specifies
a
total
bacteria
count
of 10
colonies/L.
Refer to
Test Method
F 488
for analysis
procedure.
7
8.4.2
Chloride
Solution,
Standard
(1.0
mL
= 1.00
mg
Chloride):
Dry
approximately 0.5
g
of
sodium
chloride
(NaCI)
for 1 h
at 100°C
and cool
in
a
desiccator. Dissolve
0.165
g
of the
dry
salt in
a
100
mL a
volumetric
flask
with
water,
and fill
to
mark
with water.
8.4.3
Fluoride
Solution,
Standard
(1.0
mL
=
1.00
mcj
Fluoride):
Dry
approximately
0.5 g
of
sodium
fluoride
(NaF)
for
1
h
at
100°C
and
cool ina
desiccator.
Dissolve
0.221
g
of the
dry
salt
in a 100
mL volumetric
flask
with
water,
and
fill to
mark
with
water.
8.4.4
Formate
Solution,
Standard
(1.0
mL
= 1.00
mq
Formate):
Dissolve
0.151
g
of
sodium
formate
in a 100
mL volumetric
flask
with
water,
and
fill
to
mark
with
water.
8.4.5
Nitrate
Solution,
Standard
(1.0
mL
= 1.00
m
Nitrate):
Dry
approximately
0.5
g
of
sodium
nitrate
(NaNO
3
)
for
48 h
at 105°C
and
cool
in
a
desiccator. Dissolve
0.137
g
of
the dry
salt
in
a 100
mL
volumetric
flask
with
water,
and fill
to
mark with
water.
8.4.6
Nitrite
Solution,
Standard
(1.0
mL
= 1.00
mq
Nitrite):
Dry
approximately 0.5
g
of
sodium
nitrite
(NaNO
2
)
for 24
h in a desiccator
containing
concentrated
sulfuric
acid.
Dissolve
0.150
g
of the
dry
salt
in
a
100
mL
volumetric
flask
with
water,
and
fill to
mark
with
water.
Store
in
a sterilized
glass
bottle.
Refrigerate
and
prepare
monthly.
Note
6:
Nitrite
is
easily
oxidized,
especially
in the presence
of moisture.
Use
only fresh
reagent.
Note 7:
Prepare
sterile
bottles for
storing
nitrite
solutions
by
heating
for 1
h at
1700C
in
an
air
oven.
8.4.7
Ortho-Phosphate Solution,
Standard
(1.0
mL
=
1.00 mg
o-Phosphate):
Dissolve
0.150
g
of
anhydrous
dibasic
sodium
phosphate
(Na
2
HPO
4
)
in
a 100
mL
volumetric
flask
with
water,
and fill
to mark
with
water.
8.4.8
Sulfate
Solution,
Standard
(1.0
mL
=
1.00
m
Sulfate):
Dry
approximately
0.5
g
of
anhydrous
sodium
sulfate
(Na
2
SO
4
)
for 1 h
at
110°C
and
cool
in a
desiccator.
Dissolve
0.148
g
of
the
dry salt
in
a 100
mL
volumetric
flask
with
water,
and fill
to
mark
with
water
8.5
Mixed
Anion
Solution,
WorkinQ:
Prepare
a 0.2
mgIL
and
at
least
3 different
working
standards
concentrations
for
the
analyte
anions
of interest
bracketing
the
desired
range
of analysis,
typically
between
0.2
and
50
mg/L,
and
add
2
mgIL
formate
to
all
standards.
Add
an
appropriate
aliquot
of Individual
Anion
Stock
Solution
(8.4)
to
a
pre-rinsed
100
mL volumetric
flask,
and dilute
to
100
mL
with
water.
Note 8: Use
100
L
of Individual
Anion
Stock
Solution
(8.4)
per 100
mL for
1 mg/L
anion.
Note
9:
Anions
of
no interest
may
be
omitted.
Note
10:
The
mid-range
Mixed
Anion
Solution,
Working
may
be
used
for
the
determination
of
migration
times
and
resolution
described
in 12.1.
8.6
Calibration
Verification
Solution
(CVS):
A
solution
formulated
by the
laboratory
of
mixed
analytes
of
known
concentration
prepared
in water.
The
CVS
solution
must
be
prepared
from
a
different
source
to
the
calibration
standards.
8.7
Performance Evaluation
Solution
(PES):
A
solution
formulated
by
an
independent
source
of
mixed
analytes
of known
concentration
prepared
in water.
Ideally,
the
PES
solution
should
be purchased
from
an
independent
source.
8
8.8
Quality
Control
Solution
(QCS):
A
solution
of
known analyte
concentrations
added
to
a
synthetic
sample matrix
such
as
Substitute
Wastewater
that
sufficiently
challenges
the
Test
Method.
8.9
Buffer
Solution
(100
mM CHES
I
1 mM
Calcium
Gluconate):
Dissolve
20.73
g
of
CHES
(2-[N-Cyclohexylamino]-Ethane
Sulfonic
Acid)
and 0.43
g
of
Calcium
Gluconate
in a 1 L
volumetric
flask
with
water, and
dilute to
1
L
with
water.
This
concentrate
may
be stored
in a capped
glass
or plastic
container
for up to
lyear.
8.10 Chromate
Concentrate
Solution
(100
mM Sodium
Chromate):
Dissolve
23.41
g
of
sodium
chromate
tetrahydrate
(Na
2
CrO
4
.4
H
2
0)
in a 1 L volumetric
flask
with
water,
and
dilute
to
1 L
with
water. This
concentrate
may be
stored
in a capped
glass
or
plastic container
for up to
1
year.
8.11
OFM
Concentrate
Solution
(100
mM Tetradecyltrimethyl
Ammonium
Bromide):
Dissolve
33.65 g
of
Tetradecyltrimethyl
Ammonium
Bromide
(TTABr)
in a 1
L
volumetric
flask with
water,
and
dilute
to 1
L with water.
Store
this
solution
in a
capped
glass
or
plastic container
for
up to
1 year.
Note
11:
TTABr
needs to
be converted
to the hydroxide
form
7
(TTAOH)
for use with this
test
method.
TTAOH
is
commercially
available
as
100 mM
TTAOH
which
is an equivalent
substitute.
8.12
Sodium
Hydroxide
Solution
(500 mM
Sodium
Hydroxide)--
Dissolve
20
gof
sodium
hydroxide
(NaOH)
in
a
1
L plastic
volumetric
flask
with
water, and
dilute to
1 L with
water.
8.13
Electrolyte
Solution,
Workin
(4.7
mM
Chromate
/
4
mM
TTAOH
/ 10 mM CHES
I
0.1 mM
Calcium
Gluconate)
: Wash
the anion
exchange
cartridge
in the
hydroxide
form (7.3)
using
the
20
mL
plastic
syringe
(7.4)
with 10
mL
of
500 mM
NaOH
(8.12)
followed
by 10 mL
of
water.
Discard
the
washings.
Slowly pass
4 mL
of
the
100
mM
TTABr
Solution
(8.11)
through
the
cartridge
into
a
100
mL
volumetric
flask.
Rinse the
cartridge
with
20 mL
of water, adding
the washing
to the
volumetric
flask.
Note 12:
The
above
procedure is
used
to convert
the
TTABr to
TTAOH,
which is used
in the
electrolyte.
If
using
commercially
available
100
mM TTAOH,
the above
conversion
step
is
not
necessary;
substitute
0.5 mL of
100
mM TTAOH
and continue
below
Into
the 100 mL
volumetric
flask
add 4.7
mL of
Chromate
Concentrate
Solution
(8.10)
and
10 mL of Buffer
solution
(8.9).
Mix
and dilute
to 100 mL
with
water.
The
natural pH
of the
electrolyte
should
be
9 ± 0.1.
Filter and
degas
using
the
vacuum
filtration
apparatus.
Store
the
any
remaining
electrolyte
in
a
capped
glass
or plastic
container
at ambient
temperature.
The
electrolyte
is
stable
for
1
year.
7) Available
from
Waters
Corp. as lonSelect
100mM
OEM
Hydroxide
Concentrate,
100
mM
TTAOH,
PIN
49387.
8)
Availiable from
Waters
Corp.
as lonSelect
High
Mobility Anion
Electrolyte,
PIN
49385.
9
9
Precautions
9.1
Chemicals used
in this
test
method
are
typical
of many
useful
laboratory
chemicals,
reagents
and
cleaning
solutions,
which
can
be
hazardous
if not
handled
properly.
Refer
to Guide
D
3856.
9.2
It is
the responsibility
of
the
user
to prepare,
handle,
and
dispose
of chemical
solutions
in
accordance with
all applicable
federal,
state,
and
local
regulations.
9.3
Warning
-- This
capillary
electrophoresis
method
uses
high
voltage
as
a
means
for
separating
the
analyte
anions,
and
can
be hazardous
if
not
used
properly.
Use
only
those
instruments
that
have
all
proper
safety
features.
10
Sampling
10.1 Collect
samples
in
accordance
with
Practice
D 3370.
10.2
Rinse
samples
containers
with
sample
and discard
to eliminate
any
contamination from
the
container.
Fill
to overflowing
and
cap
to exclude
air.
10.3 Analyze
samples
as
soon as
possible
after
collection.
For
nitrite,
nitrate,
and
phosphate
refrigerate the
sample
at
4°C
after
collection.
Warm
to room
temperature before
dilution
and
analysis.
10.4
At the
lab, filter
samples
containing
suspended
solids
through
a
pre-rinsed
0.45
pm
aqueous
compatible
membrane
filter
before
analysis.
10.5
If sample
dilution
is
required
to remain
within
the
scope
of
this
Test Method,
dilute
with water
only.
11
Preparation
of Apparatus
11.1
Set up
the
CE
and
data
system
according
to
the manufacturer’s
instructions.
11
2
Program
the CE
system
to maintain
a
constant
temperature
of 25°
±
0.5°C;
or
5°C
above
ambient
laboratory
temperature.
Fill
the
electrolyte
reservoirs
with
fresh
chromate
electrolyte
working
solution
(8.13),
and
allow
10 mm
for
thermal
equilibration.
11.3
Condition
a
new
capillary
(7.1.6)
with
500 mM
NaOH
Solution
(8.12)
for
5 mm
followed
by
water
for
5
mm.
Purge the
capillary
with
electrolyte
(8.13)
for
3
mm.
11.4
Apply
15 kV
of voltage
and
test for
current.
The current
should
be 14±
1
pA.
If
no
current
is
observed,
then
there
is
a
bubble
and/or
blockage
in the
capillary.
Degas
the
chromate
electrolyte
working
solution
and
retry.
If still
no current,
replace
the
capillary.
11.5
Set
the
UV detector
to
254
nm detection,
or equivalent.
Zero the
detector
to
0.000
absorbance.
UV offset
is
less then
0.1 AU.
11.6
Program
the CE
system
for
constant
current
of 14
pA.
11.7
Program
the
CE
system
for
a hydrostatic
sampling
of
30
s. Approximately
37nL
of
sample
is
siphoned
into
the
capillary.
Different
sampling
times
may
be
used
provided
that
the
samples
and
standards
are
analyzed
identically.
11.8
Program
the
CE
system
for
a 1 mm
purge
with
the
chromate
electrolyte
working
solution
between
each
analysis.
Using
a
15
psi
vacuum
purge
mechanism,
one
60
cm
capillary
volume
can
be
displaced
in
30
s.
10
11.9
Program
the
data system
for an
acquisition
rate
of
at
least
20
points
per
s.
Program
the data
system to
identify
analyte
peaks based
upon
normalized
migration
time
using
Cl as
the reference
peak,
and
to quantitate
analyte
peak
response
using
time
corrected
peak
area.
Note 13: Under
the analysis
conditions
Cl
is always the
first
peak
in
the
electropherogram,
and
can be
used as
a
migration time
reference
peak.
12
Calibration
12.1
Determination
of
Migration
Times--
Calibrate
Daily.
The migration
time
of an
anion is
dependent
upon
the
electrolyte
composition,
pH, capillary
surface
and
length,
applied
voltage, the
ionic
strength
of the
sample,
and
temperature.
For
every
fresh
electrolyte
determine
the analyte
migration
time, in mm
to
the
third
decimal
place,
of the
mid-range
mixed anion
standard
working
solution
(8.5),
described
in
Sec
11. Use
the
mid-point
of analyte
peak
width as the
determinant
of
analyte
migration
time.
Note 14:
Analyte
peak apex
may
be
used as
the
migration
time
determinant,
but potential
analyte
misidentification
may
result
with
asymmetrical
peak
shape at high
analyte
concentrations.
12.2 Analyze
the blank
(8.3),
a 0.2
mg/L,
and
at
least
3
working
mgIL
solutions
(8.5),
using the
set-up
described
in
sec
11. For each
anion
concentration
(X-axis)
plot
time
corrected
peak
area
response
(Y-axis).
Determine
the best
linear
calibration
line
through
the
data
points,
or
use
the
linear
regression
calibration
routine
(1/X
Weighting
and Linear
Through
Zero) available
in
the
data system.
Note
15:
Do
not
use peak
height
for
calibration.
Peak
area is
directly related
to migration
time,
i.e.
for the
same analyte
concentration,
increasing
migration
time gives
increasing
peak area.
Note
16:
EPA recommends
calibration
at the
minimum
concentration
of
0.2 mgIL
plus
3 additional
points.
The
r
2
(coefficient
of determination)
values
should be
greater
than
0.995;
typical
r
2
values
obtained
from
the interlaboratory
collaborative
are
given
in
Table
A2.
12.3 Calibrate
daily
and
with each
change
in
electrolyte,
and
validate
by
analyzing
the
CVS
solution
(8.6)
according
to procedure
in
Seci 6.4.
12.4 After
validation
of
linear multiple
point
calibration,
a single
point calibration
solution
can be
used between
0.2 and
50
mgJL
for
recalibration
provided
the
quality
control
requirements
in
Sec
16.4
are
met.
13
Procedure
13.1 Dilute
the
sample,
if necessary
with
water, to
remain within
the scope
(Sec
1.2,
1.3)
and calibration
of this
test method.
Refer
to
Al .5.1.
13.2
Analyze
all
blanks
(8.3),
standards
(8.5),
and
samples
as described
in
Sec
11
using
the
quality control
criteria
described
in
Sec 16.5
to 16.9.
Refer to
Fig.
1
through
5
for
representative
anion
standard,
detection
limit standard,
substitute
wastewater,
drinking
water, and
wastewater
electropherograms.
13.3 Analyze
all blanks,
calibration
standards,
samples,
and quality
control
solutions
in
singlicate.
Perform
at
least
one
matrix
spike analysis
in
duplicate
as
part
of
the
QC
protocol,
Sec 16.7.
Optional:
Duplicate
analyses
are
preferred
due
to
short
analysis
times.
Note 17: Collaborative
data
was
acquired, submitted
and
evaluated
as
the average
of
duplicate
samplings.
11
13.4
After 20
sample
analyses,
or
batch, analyze
the
QCS
solution
(8.8). If
necessary,
recalibrate
using
a single
mixed
anion standard
working
solution
(8.5),
and
replace
analyte
migration
time.
Note
18: A
change
in analyte
migration
time of the
mixed anion
standard working
solution
by more
than
+5%
suggests
that
components
in
the
previously
analyzed
sample
matrices
have
contaminated
the
capillary
surface.
Refer
to
sec
6.7.
Continue
but
wash the capillary
with
NaOH
solution
(8.12)
before
the next
change in
electrolyte.
14
Calculation
14.1 Relate
the
time corrected
peak
area response
for
each analyte
with the
calibration
curve generated
in section
12.2 to
determine
mg/L
concentration
of
analyte
anion.
If the sample
was
diluted
prior to
analysis,
then
multiply
mg/L
anion by
the
dilution
factor to obtain
the
original
sample
concentration,
as
follows:
Original
Sample
mg/L
Analyte =
(A
x SF)
where;
A
= analyte
concentration
determined
from
the
calibration
curve,
in
mg/L,
SF
= scale
or dilution
factor.
15 Report
Format
15.1 The
sample
analysis
report should
contain the
sample
name,
analyte
anion
name,
migration
time
reported
to
3 decimal
places,
migration
time
ratio,
peak
area,
time
corrected
peak
area,
sample
dilution,
and
original solution
analyte
concentration.
Optional:
Report analysis
method
parameters,
date of
sample
data
acquisition,
and
date
of
result
processing
for
documentation
and validation
purposes.
16
Quality
Control
16.1 Before
this
test
method
is
applied
to the
analysis
of unknown
samples,
the
analyst
should
establish
quality
control according
to
procedures
recommended
in
Practice
D5847, and
Guide D5810.
16.2 The
laboratory
using
this
test must
perform an
initial
demonstration
of
laboratory
capability
according
to
procedures
outlined
in Standard
Practice
D5847,
and
Appendix
C.
Note
19:
Certified
Performance
Evaluation
Solutions
(PES)
and
QC
Solutions
(QCS and
CVS)
are
commercially
available,
and
recommended.
16.3
Initial
Demonstration
of Performance:
Analyze
seven
replicates
of a
Performance
Evaluation
Solution
(PES,
8.7).
Calculate
analyte
concentration
mean
and
standard
deviation
of the
seven replicates
and compare
to
the
precision
and
Initial
%Recovery
for the
analyte
in reagent
water
given
in
Table
8.
16.3.1
Repeat
the
7 replicate
analysis
protocol before
using a
freshly
prepared
QVS
solution
(8.6)
and
QCS
solution
(8.8) for
the first
time.
Calculate
the
standard
deviation
and
compare
with
previous
results
using the
student
t-test.
If no
significant
difference
is
noted then
use the
combined
standard
deviation
to
determine
the
QC
limits,
for
the
QVS
and
QCS
solutions.
16.4
Calibration
Verification:
After
calibration,
verify
the
calibration
linearity
and
acceptable
instrument
performance
using a Calibration
Verification
Solution
(8.6)
treated
as an
unknown.
If the
determined
CVS
concentrations
(8.6)
are
not
within
± 3
standard
deviations
of the known
true
values
as
described
in 16.3.1,
the
calibration
solutions
may be
out of control.
Reanalyze,
and
if analyte
concentration
still falls
outside
the
acceptable
limits,
fresh calibration
solutions
(8.5)
are
required.
Successful
CVS
analyte
concentration
must
be
confirmed
after
recalibration
before
continuing
with the
Test
Method.
12
16.5
Analyze
a
reagent
blank
(8.3)
with each
batch
to check
for
contamination
introduced
by
the laboratory
or
use
of the Test
Method.
16.6
Quality
Control
Solution:
Analyze
one
QCS
(8.8)
after
20 samples,
or batch.
The
analyte
concentrations
for the
QCS
should
fall within
the
lower limit
(LL)
and
upper
limits
(UL)
given
in Table
8.
16.7
Matrix
Spike
Recovery:
One Matrix
Spike (MS)
must be
analyzed
in
duplicate
with
each
batch
of
samples
to test method
recovery
and
relative
%difference
between
them.
Spike
a
portion
of one
sample
from each
batch
with
a
known
concentration
of analyte,
prepared
in accordance
with
Guide
D3856. The
%
recovery
of the spike
should
fall
within the
MS/MSD
lower
and
upper limits,
and
the
Relative
%Difference
given
in
Table
8
for
the appropriate
sample
matrix.
If it
does
not,
an
interference
may
be
present
and the
data
for the set
of similar
samples
matrices
must
be qualified
with
a warning
that the
data
are suspect,
or
an alternate
test method
should be
used. Refer
to
Guide
D581
0.
16.7.1 If the
known
analyte
concentration
is between
15
and
50 mg/L,
then
spike the
sample
solution
to increase
analyte
concentration
by 50%.
16.7.2
If
the
known analyte
concentration
is
between
2 mg/L
and 15
mgJL,
then
spike
the
sample
solution
to increase
analyte
concentration
by 100%,
but
not
less than
2
mg/L.
16.7.3 If the
known
analyte
concentration
is
less than
2 mgIL,
then
spike the
sample
solution
with 1 mgIL,
5
times
the ML.
16.7.4
Calculate
the
percent
recovery of
the spike
using
the
following
formula:
%
Recovery
=
100
[A (Vs+V)
- B
Vs]/C
V
where
A
= Analyte
Concentration
(mgIL) in
Spiked Sample
B
= Analyte
Concentration
(mgIL)
in Unspiked
Sample
C
=
Concentration
(mgIL)
of
Analyte
in Spiking
Solution
V
= Volume
(mL)
of
Sample
Used
V
= Volume
(mL) Added
with Spike.
Evaluate
performance
according
to
Practice
D5847.
16.8
Method
Precision:
One
unknown
sample should
be analyzed
in triplicate
with
each
batch
to test method
precision.
Calculate
the
standard deviation
and
use
the F-Test
to
compare
with the
single operator
precision
given
in Tables
1
through
7
for
the
equivalent
analyte
concentration
and
matrix
type.
Evaluate
performance
according
to
Practice
D5847.
16.9
The
laboratory
may perform
additional
quality control
as
desired
or
appropriate.
17 Precision
and
Bias
17.1 The
precision
and bias
data presented
in
this
test
method
meet the
requirements
of Practice
2777-98,
and are
given in
Tables
1 through 7.
The
full
Research
Report,
RR#
Dl 9-1165,
can
be
obtained
from
ASTM
Headquarters.
17.2
This
test
method
interlaboratory
collaborative
was performed
by
11
laboratories
using
one
operator
each. Four
Youden
Pair spike
concentrations
for the
7
analytes
anions yielding
8
analyte
concentration
levels.
Test data
was
submitted
for 11
Reagent
Waters,
11 Substitute
Wastewaters,
15
Drinking
Waters,
and 13
Wastewater
sample
matrices.
13
17.3
All
data
given
in
this method
was
quantitated
using
non-weighted
linear
calibration
through
zero,
except
where
noted.
17.4 The
precision,
bias,
and
matrix recovery
of this test method
per anion
analyte
in
the 4 tested
sample
matrices are
based upon
the
analyte
true value,
calculated
using
weight, volume, and
purity.
True
value spiking
solution
concentrations
are
given in
Table A4.
17.5
The bias and
matrix recovery
statements
for less than
2 mglL of chloride,
sulfate,
and
nitrate in
naturally
occurring
sample
matrices
maybe misleading
due to
spiking
of small analyte
concentration
into a high naturally
occurring
analyte
concentration
observed
with
the
matrix
blank.
The
commonly occurring
analyte
concentrations
observed
in
the sample
matrix blanks for
the naturally occurring
tested
matrices
are given
in Table A5.
17.6 The high
nitrate bias and
%recovery
noted
for the
0.5 mgIL
NO
3spike solution
are
attributed to the
spiking solution
containing
50
mgIL
nitrite
and
0.5
mg/L
nitrate.
Refer
to Appendix
Table A4, Solution
3.
Some
of the
nitrite
converted
to
nitrate prior
to analysis. Similar
NO
conversion
effect
is
observed
with the 2
mgIL
nitrate and 2 mg/L nitrite
spike, Solution
7.
17.7
All
collaborative participants
used the
premade
Chromate electrolyte,
(lónSelect
High Mobility Anion
Electrolyte,
available from
Waters
Corp.) Ten
laboratories
used
a
Waters
CIA Analyzer
with Millennium
Data Processing
Software,
and one
laboratory used a
Agilent CE
System with
Diode Array
Detector
that provided
equivalent
results,
although
different
sampling
and
detection
conditions
were
necessary
for equivalent performance.
Note 20:
Refer to reference Bi
.16 and Agilent
(the former HP
Company) website
for
recommended
operating conditions.
18
Key
Words
Anion
Capillary Electrophoresis
Drinking
Water
Ion Analysis
Reagent Water
Substitute Wastewater
Wastewater
14
Appendix
A
Mandatory
Information
Al .1 All data
presented
in the
following
Tables
conform
and
exceed the
requirements
of
D2777-98.
Data
from
eleven
reagent
waters,
eleven
substitute
wastewater,
fifteen
Drinking
Water,
and
thirteen
wastewater
sample
matrices,
were
tested using
a set
of 4 Youden
Pair
concentrations
for 7
analyte anions.
All
submitted
individual
data
points
are the average
of
duplicate
samplings.
Al .2 Calibration
Linearity
Al
.2.1 All laboratories
used
a provided
set
of 4
certified, mixed
anion
calibration
solutions
in
concentrations
between
0.5
mg/L and
50
mgIL,
formulated
in
random concentrations
given
in Table
Al.
They were
prepared
from
certified,
individual
1000 mg/L
Stock
Standards
obtained
from
APG,
lnc, Beipre,
Ohio.
No
dilution was
necessary.
Table
Al:
Collaborative
Calibration
Standard,
mpIL
Concentrations
Analyte
Anion
Standard
1
Standard
2
Standard
3
Standard
4
Chloride
50
25
0.5
10
Bromide
0.5
25
10
50
Nitrite
25
0.5
50
10
Sulfate
10
25
0.5
50
Nitrate
25
0.5
50
10
Fluoride
5
0.5
10
25
Phosphate
50
25
0.5
10
Al .2.2
A
Linear Through
Zero; no
weighting
regression
was
used
to calculate
the
calibration
curve.
The
range coefficient
of determination
(r
2)
values
obtained
from
the
collaborative
is
shown in Table
A2
Table
A2: Expected
Rancje
of
(r
2)
Coefficient
of
Determination
Anion
I
r’
Average,
n29
Lowest
Highest
Chloride
0.99987
0.99959
0.99997
Bromide
0.99953
0.99878
0.99996
Nitrite
0.99983
0.99961
0.99999
Sulfate
0.99976
0.99901
0.99999
Nitrate
0.99957
0.99840
0.99999
Fluoride
0.99972
0.99797
0.99999
Phosphate
0.99982
0.99942
0.99999
Al
.2.3 EPA
requires
that
1/X
weighting
be used
for
calibration.
The
P
&
B
data
were derived
using
unweighted
calibration.
Table
A2a shows
there
is no
significant
difference
in r
2 linearity
between
these
2
calibration
routines.
Table
A2a
Coefficient
of
Determination
r
2
from
a Single
Calibration
Analyte
No Weighted
lIx
Weighted
Anion
Calibration
Calibration
Chloride
0.99994
0.99996
Bromide
0.99942
0.99923
Nitrite
0.99975
0.99981
Sulfate
0.99971
0.99974
Nitrate
0.99975
0.99974
Fluoride
0.99986
0.99967.
Phosphate
0.99999
0.99999
15
Al .3
Quality
Control
Solution
Preparation
Al
.3.1 The
Quality Control
Solution
(QCS)
was
also used
as the
Calibration
Verification
Solution (CVS).
Al
.3.2
Quality
Control Solution
(QCS)
was manufactured,
analyzed using ion
chromatography,
and
certified by APG
as 1 OOX
concentrate, to replicate typical
Drinking
Water concentrations. Required 1:100
dilution with water before
analysis.
The
QCS
analyte
concentrations,
required
control limits, and
interlaboratory
determined control
limits based
upon n# analyses are
given in
Table A3.
Table A3:
Quality
Control Acceptance
Limits
Analyte
True Value
Certified
Required
Determined
Anion
mg/L
Value
99%
QCS
mgJL
Confidence
Mean ± Std Dev,
Interval
n=82
Chloride
48.68
48.61
± 0.12
43.99—
52.96
47.64±1.53
Bromide
0.00
0.00
0.00
0.00
Nitrite
2.87
2.90
± 0.07
2.39—
3.26
2.88 ± 0.19
Sulfate
35.69
35.63 ± 0.25
29.54
— 40.53
35.02
± 1.21
Nitrate
15.76
15.78 ±
0.15
12.80—
18.39
15.33 ± 4.35
Fluoride
1.69
1.68 ± 0.01
1.49
— 1.87
1.67 ± 0.09
Phosphate
5.47
5.55
±
0.12
4.78—
6.20
5.58 ±
0.28
Al
.3.3
A
single day’s
QCS
was reprocessed
using
a 1IX weighting linear
calibration
and remained within the
QC
Acceptance
Limits.
Table A3a QC
Standard Results:
Reprocessed
Using
lIx Calibration
Analyte
No
Weighted
lIx Weighted
QC
Acceptance
Anion
Calibration
Calibration
99%Conf Interval
Chloride
48.64
± 1.06 48.77
± 1.07
43.99
— 52.96
Nitrite
2.93 ± .03
2.82
± .03
2.39
— 3.26
Sulfate
34.49 ± .79
34.64
± .79
29.54
— 40.53
Nitrate
15.28±.15
15.23±.18
12.80—18.39
Fluoride
1.74
± .02
1.63
± .02
1.49
— 1.87
Phosphate
5.75 ± .15
5.77
± .15
4.78— 6.20
Al .4
Youden
Pair
Spiking
Solution
Preparation
Al .4.1
Eight mixed
anion, 1
OOX concentrate,
spiking
solutions
were
prepared
in
accordance
with
Sec 8.3 (Reagents and
Materials)
of the test
method
using
anhydrous
sodium salts.
The mg/L
concentrations
of
the
eight
standards
followed the
approved Youderi
Pair design
- 0.5 & 0.7, 2
& 3,
15
& 20,
40 &
50
mg/I
for all anions
except fluoride,
which
is
0.5 &
0.7, 2
& 3, 7&
10, 20
&
25mgIL.
The analyte
true
value
concentrations
were randomized
among
the
eight
spiking solutions
as
described in Table
A4.
Al .4.2
A
ninth
solution containing
approximately
10
mg/L
of each
analyte was
diluted
1:50
with
water, and
was used for
method
detection
limit
calculations.
16
Table A4:
True Value Youden Pair
Spiking mci/L
Concentrations
Anion/TV
1
2
3
4
5
6
7
8
9
Chloride
0.71
2.00
2.98
14.92
39.81
19.91
49.76
0.50
10.20
Bromide
2.00
3.01
14.93
39.81
19.91
49.77
0.70
0.51
10.49
Nitrite
2.98
39.61
19.81
14.86
49.52
0.50
.2.00
0.70
9.94
Sulfate
39.60
49.51
0.49
0.70
1.98
2.98
14.86
19.81
10.23
Nitrate
14.92
19.19
39.87
49.78
0.50
0.70
2.00
2.98
10.35
Fluoride
2.00
0.71
0.50
3.00
9.99
6.99
19.98
24.99
10.40
Phosphate
49.51
39.60
19.90
0.50
2.98
1.99
0.69
14.86
10.48
These
solutions, kept at ambient
temperature, were analyzed before and during
the
collaborative
to monitor for
accuracy
and
stability. The
mg/L
True Value
in
was used to
determine
bias, matrix recovery,
and
the
single
operator
and
interlaboratory
precision in
the P
&
B tables per the requirement of D
2777.
Solution
3
and
7 exhibited some conversion
of nitrite to nitrate before
analysis.
This conversion
is evident in the
bias and % Recovery for
0.5
mgIL
and 2
mg/I
nitrite and
nitrate.
Al
.5
Sample
Matrix
Preparation
Al .5.1 All
participating laboratories provided
and tested reagent water,
substitute
wastewater,
naturally occurring drinking
water, and naturally
occurring
wastewater. Before
matrix spiking
with the Youden Pair
solutions, the sample
matrix was evaluated, then appropriately
diluted to give the highest anion
concentration
below
50 mg/L.
The
diluted sample matrix
was used
to
dilute
each
Youden Pair spiking solution
1:100.
Al
.5.2
Reagent Water was used as-is.
Substitute wastewater
was
diluted
1:20
with
water. Naturally occurring drinking water
was
used as-is or diluted
1:5
with
water.
Naturally occurring wastewater
was diluted between
1:3 and 1:20,
except
one
which required
a 1:1000 dilution due
to high
chloride.
Al
.5.3
Due to the anion content
of
the
naturally occurring
drinking
water
and “real”
wastewater
matrices,
some of the reported
spike matrix results exceeded
the
scope of this
test
method. Linearity and matrix
recovery data obtained
from
the
collaborative indicated that these
data are acceptable,
and
extended
the
useful
range
of this test method.
Al .5.4
Due to the anion content
of
the
naturally occurring
sample matrices
given
in Table A5,
the low
concentration bias and
recovery may
be
misleading
because of spiking a low anion
concentration
increment into a large
naturally
occurring concentration of the same
anion.
Table
A5: Blank
Analyte
Concentrations for
Naturally
Occurring Sample
Matrices
Data
in mgIL
Chloride
Sulfate
Nitrate
Drinking
Water
0.7
to
41.9
0.5
to
33.6
0.2
to
6.5
Substitute
Wastewater
20.5 to 25.5
3.2 to 4.0 Not Detected
“Real”
0.9
to 43.4
0.5 to 50.4
0.3
to
23.0
Wastewater
17
Al
.6
Test
Method
Detection
Limits:
Al
.6.1
Spiking
Solution #9,
containing
10 mgIL of each
analyte,
was
diluted
1:50 with
water
and
was
used
for
detection
limit calculations.
Ten laboratories perlormed
seven
replicate
samplings,
and
the mean and
standard
deviation from each
laboratory
was
calculated.
The
mean time corrected
peak area
response
for the
7
replicates was
given
the true value of
the solution #9, and
from a simple
proportion,
the
standard
deviation was calculated
as
mg/L.
Std Dev,
mg/L
= (True
Value
Conc Sol’n #9,
mq/L)(Response
Std Dev)
Ave Response
of SoI’n
#9
Al
.6.2
Method
detection
limits (MDL) were
derived
using
“pooled”
EPA protocol
and
the
student
t-test at 6
degrees of
freedom, as follows;
The
method
detection
limit (MDL)
=(3.14)(Std
Dev,
mg/L).
Al
.6.3
The
upper
and
lower
confidence
limits
were calculated as;
95%
Confidence
Interval:
LCL
(Lower
Confidence Limit)
= 0.64
x
MDL
UCL
(Upper
Confidence Limit)
= 2.20
x MDL
Al
.6.4
Method
Detection
Limits
are given
in
Table
A6.
Table
A6: Method Detection
I imif
Anion
mgIL
Solution
Method
Detection I
95% Confidence
Interval
Concentration
MDL,_mgIL
mg/L
Chloride
0.204
0.075
0.048
to
0.165
Bromide
0.210
0.120
0.077
to
0.264
Nitrite
0.199
0.103
0.066
to 0.227
Sulfate
0.205
0.065
0.042
to 0.143
Nitrate
0.207
0.076
0.049
to
0.167
Fluoride
0.208
0.032
0.020
to 0.070
Phosphate
0.210
0.097
0.062
to 0.213
18
Table
1
Precision,
Bias,
and
Matrix Recovery
for
Chloride
Matrix
# of
True
Mean
Bias
vs
Recovery
Interlab
Interlab
Single
Analyst
Values
Value
Result
True
vs True
Std Dev
%RSD
Operator
%RSD
Value
Value
S(t)
Std Dev,
S(o)
Reagent
9
0.50
0.55
0.05
110.0
0.11
19.8
Water
10
0.71
0.69
-0.02
97.2
0.08
11.5
0.05
7.5
10
2.00
1.97
-0.03
98.5
0.14
6.8
9
2.98
2.97
-0.01
99.7
0.11
3.8
0.05
2.1
10
14.92
14.76
-0.16
98.9
0.61
4.2
10
19.91
19.81
-0.10
99.5
0.81
4.1
0.48
2.8
10
39.81
38.58
-1.23
96.9
1.43
3.7
10
49.76
48.70
-1.06
97.9
1.94
4.0
1.36
3.1
Substitute
9
0.50
0.46
-0.04
92.0
0.51
111.1
Wastewater
9
0.71
0.43
-0.28
60.6
0.69
160.7
0.42
93.8
9
2.00
1.52
-0.48
76.0
0.68
45.0
9
2.98
2.58
-0.40
86.6
0.63
24.5
0.50
24.3
9
14.92
14.29
-0.63
95.8
1.02
7.1
9
19.91
18.93
-0.98
95.1
1.24
6.6
0.60
3.6
9
39.81
37.34
-2.47
93.8
5.44
14.6
9
49.76
47.54
-2.22
95.5
3.13
6.6
4.43
10.4
Drinking
12
0.50
0.63
0.13
126.0
0.67
106.1
Water
12
0.71
0.75
0.04
105.6
0.34
45.5
0.40
57.2
12
2.00
2.15
0.15
107.5
0.51
23.6
12
2.98
2.95
-0.03
99.0
0.39
13.1
0.47
18.5
12
14.92
14.54
-0.38
97.5
0.71
4.9
12
19.91
19.09
-0.82
95.9
1.11
5.8
0.37
2.2
12
39.81
38.38
-1.43
96.4
1.56
4.1
49.76
47.97
-1.79
96.4
2.19
4.6
1.26
3.9
ReaI
9
0.50
0.42
-0.08
84.0
0.34
81.0
Wastewater
10
0.71
0.47
-0.24
66.2
0.34
72.6
0.26
59.3
10
2.00
1.56
-0.44
78.0
0.51
32.7
9
2.98
2.78
-0.20
93.3
0.19
6.8
0.37
17.3
10
14.92
14.29
-0.63
95.8
0.63
4.4
10
19.91
18.83
-1.08
94.6
0.78
4.1
0.46
2.8
9
39.81
37.01
-2.80
93.0
2.78
7.5
10
49.76
48.24
-1.52
96.9
3.15
6.5
2.54
6.0
19
Table 2
Precision, Bias, and Matrix Recovery for Bromide
Matrix
# of
True
Mean
Bias
vs
Recovery
Interlab
lnterlab
Single
Analyst
Values
Value
Result
True
vs True
Stcf Dev
%RSD
Operator
%RSD
Value
Value
S(t)
Std
Dev, S(o)
Reagent
10
0.51
0.60
0.09
117.6
0.19
31.0
Water
10
0.70
0.83
0.13
118.6
0.23
28.2
0.10
14.6
10
2.00
2.06
0.06
103.0
0.14
6.6
10
3.01
2.88
-0.13
95.7
0.23
7.9
0.15
6.3
10
14.93
15.00
0.07
100.5
0.58
3.9
10
19.91
19.32
-0.59
97.0
0.97
5.0
0.75
4.4
10
39.81
39.66
-0.15
99.6
1.24
3.1
10
49.77
50.04
0.27
100.5
2.94
5.9
1.61
3.6
Substitute
9
0.51
0.67
0.16
131.4
0.19
28.8
Wastewater
9
0.70
0.96
0.26
137.1
0.21
21.8
0.08
9.3
9
2.00
2.14
0.14
107.0
0.22
10.2
9
3.01
2.72
-0.29
90.4
0.35
12.8
0.17
7.0
9
14.93
14.70
-0.23
98.5
0.58
3.9
9
19.91
18.91
-1.00
95.0
2.62
13.8
1.63
9.7
9
39.81
38.76
-1.05
97.4
1.11
2.9
9
49.77
48.81
-0.96
98.1
1.52
3.1
0.48
1.1
Drinking
13
0.51
0.58
0.07
113.7
0.25
43.4
Water
13
0.70
0.83
0.13
118.6
0.22
26.5
0.14
19.9
13
2.00
1.98
-0.02
99.0
0.25
12.5
13
3.01
2.56
-0.45
85.0
0.25
9.7
0.15
6.8
13
14.93
14.63
-0.30
98.0
0.50
3.4
13
19.91
19.22
-0.69
96.5
1.10
5.7
0.77
4.6
13
39.81
38.97
-0.84
97.9
1.99
5.1
13
49.77
48.74
-1.03
97.9
1.49
3.1
1.13
2.6
Real”
11
0.51
0.59
0.08
115.7
0.11
19.3
Wastewater
12
0.70
0.78
0.08
111.4
0.19
24.4
0.10
14.0
11
2.00
2.08
0.08
104.0
0.13
6.3
12
3.01
2.70
-0.31
89.7
0.41
15.1
0.27
11.5
12
14.93
15.16
0.23
101.5
0.90
6.0
11
19.91
19.46
-0.45
97.7
1.63
8.4
1.09
6.3
12
39.81
40.24
0.43
101.1
2.27
5.7
12
49.77
49.97
0.20
100.4
2.52
5.0
0.91
2.0
20
Table
3
Precision,
Bias, and
Matrix
Recovery
for Nitrite
Matrix
# of
True
Mean
Bias
vs
Recovery
Interlab
Intertab
Single
Analyst
Values
Value
Result
True
vs
True
Std
Dev
%RSD
Operator
%RSD
Value
Value
S(t)
Std
Dev, S(o)
Reagent
9
0.50
0.62
0.12
124.0
0.16
26.1
Water
9
0.70
0.72
0.02
102.9
0.08
10.5
0.05
7.1
10
2.00
1.31
-0.69
65.5
0.25
19.2
10
2.98
3.11
0.13
104.4
0.17
5.4
0.13
6.0
10
14.86
14.70
-0.16
98.9
0.47
3.2
10
19.81
19.88
0.07
100.4
0.70
3.5
0.27
1.5
10
39.61
39.90
0.29
100.7
0.88
2.2
10
49.52
48.24
-1.28
97.4
1.34
2.8
1.25
2.8
Substitute
9
0.50
0.37
-0.13
74.0
0.22
59.7
Wastewater
9
0.70
0.59
-0.11
84.3
0.28
48.1
0.21
43.2
10
2.00
1.25
-0.75
62.5
0.38
30.8
9
2.98
2.62
-0.36
87.9
0.82
31.4
0.43
22.1
9
14.86
14.40
-0.46
96.9
0.58
4.0
10
19.81
19.50
-0.31
98.4
1.66
8.5
0.81
4.8
10
39.61
39.97
0.36
100.9
2.02
5.0
9
49.52
49.09
-0.43
99.1
3.03
6.2
2.11
4.7
Drinking
11
0.50
0.52
0.02
104.0
0.08
14.4
Water
12
0.70
0.74
0.04
105.7
0.17
23.3
0.09
13.5
12
2.00
1.30
-0.70
65.0
0.21
15.9
12
2.98
2.97
-0.01
99.7
0.14
4.6
0.16
7.4
11
14.86
14.60
-0.26
98.3
0.40
2.8
11
19.81
19.82
0.01
100.1
0.59
3.0
0.26
1.5
11
39.61
39.35
-0.26
99.3
0.99
2.5
12
49.52
49.14
-0.38
99.2
1.93
3.9
0.64
1.5
“Real”
9
0.50
0.55
0.05
110.0
0.13
24.5
Wastewater
10
0.70
0.73
0.03
104.3
0.24
32.9
0.07
10.8
9
2.00
1.27
-0.73
63.5
0.18
14.2
10
2.98
2.99
0.01
100.3
0.19
6.2
0.15
7.0
10
14.86
14.55
-0.31
97.9
0.46
3.1
10
19.81
19.68
-0.13
99.3
0.71
3.6
0.38
2.2
9
39.61
39.21
-0.40
99.0
1.03
2.6
9
49.52
47.27
-2.25
95.5
3.50
7.4
2.40
5.6
21
Table 4
Precision,
Bias, and Matrix
Recovery for Sulfate
Matrix
# of
True
Mean
Bias vs
Recovery
Interlab
lnterlab
Single
Analyst
Values
Value
Result
True
vs True
Std
0ev
%RSD
Operator
%RSD
Value
Value
S(t)
Std Day,
S(o)
Reagent
9
0.49
0.49
0.00
100.0
0.18
37.5
Water
10
0.70
0.71
0.01
101.4
0.20
29.2
0.05
8.3
10
1.98
2.04
0.06
103.0
0.19
9.7
10
2.98
3.09
0.11
103.7
0.24
7.9
0.06
2.5
10
14.86
14.67
-0.19
98.7
0.57
4.0
10
19.81
19.67
-0.14
99.3
0.73
3.8
0.44
2.6
10
39.60
39.66
0.06
100.2
0.92
2.4
10
49.51
49.27
-0.24
99.5
1.26
2.6
0.49
1.1
Substitute
9
0.49
0.38
-0.11
77.6
0.25
66.9
Wastewater
9
0.70
0.51
-0.19
72.9
0.08
16.4
0.18
39.3
9
1.98
1.83
-0.15
92.4
0.29
16.2
9
2.98
2.86
-0.12
96.0
0.31
11.2
0.20
8.6
9
14.86
14.19
-0.67
95.5
1.06
7.7
9
19.81
19.23
-0.58
97.1
0.97
5.2
0.46
2.8
9
39.60
38.45
-1.15
97.1
1.33
3.6
9
49.51
47.75
-1.76
96.4
1.43
3.1
0.75
1.8
Drinking
12
0.49
0.41
-0.08
83.7
0.21
52.8
Water
12
0.70
0.41
-0.29
58.6
0.20
50.3
0.14
34.3
13
1.98
1.77
-0.21
89.4
0.53
30.3
13
2.98
2.68
-0.30
89.9
0.42
16.2
0.27
12.1
13
14.86
14.25
-0.61
95.9
1.11
8.0
12
19.81
19.31
-0.50
97.5
1.39
7.4
1.48
8.9
12
39.60
38.58
-1.02
97.4
1.96
5.2
13
49.51
48.43
-1.08
97.8
2.04
4.3
1.44
3.3
ReaI
10
0.49
0.37
-0.12
75.5
0.39
106.4
Wastewater
11
0.70
0.16
-0.54
22.9
1.19
765.2
0.47
179.6
11
1.98
1.57
-0.41
79.3
0.87
55.4
11
2.98
2.53
-0.45
84.9
0.64
25.4
0.24
11.9
11
14.86
14.69
-0.17
98.9
1.26
8.6
10
19.81
19.38
-0.43
97.8
0.90
4.6
0.57
3.4
11
39.60
38.74
-0.86
97.8
1.71
4.4
10
49.51
48.36
-1.15
97.7
1.51
3.1
0.47
1.1
22
Table
5
Precision,
Bias, and Matrix
Recovery
for
Nitrate
Matrix
#
of
True
Mean
Bias vs
Recovery
lnterlab
Interlab
Single
Analyst
Values
Value
Result
True
vs
True
Std
0ev
%RSD
Operator
%RSD
Value
Value
S(t)
Std Dev,
S(o)
Reagent
10
0.50
1.02
0.52
204.00
0.08
7.4
Water
10
0.69
0.71
0.02
102.90
0.08
11.6
0.06
6.4
11
1.99
2.83
0.84
142.21
0.23
8.1
11
2.97
2.89
-0.08
97.31
0.18
6.4
0.14
5.0
11
14.91
14.77
-0.14
99.06
0.44
3.0
11
19.18
19.77
0.59
103.08
0.64
3.2
0.24
1.4
10
39.86
39.09
-0,77
98.07
1.43
3.7
10
49.77
48.93
-0.84
98.31
1.72
3.5
0.62
1.4
Substitute
11
0.50
1.18
0.68
236.00
0.41
34.9
Wastewater
10
0.69
0.55
-0.14
79.71
0.30
55.3
0.42
4.9
10
1.99
2.70
0.71
135.68
0.42
15.4
10
2.97
2.33
-0.64
78.45
1.10
47.3
0.39
15.4
9
14.91
14.29
-0.62
95.84
0.78
5.4
10
19.18
18.69
-0.49
97.45
1.46
7.8
0.25
1.5
11
39.86
37.70
-2.16
94.58
1.93
5.1
11
49.77
47.78
-1.99
96.00
2.18
4.6
1.62
3.8
Drinking
11
0.50
1.06
0.56
212.00
0.19
18.1
Water
11
0.69
0.65
-0.04
94.20
0.06
8.7
0.12
14.4
12
1.99
3.05
1.06
153.27
0.39
12.8
11
2.97
3.01
0.04
101.35
0.22
7.2
0.33
10.8
12
14.91
14.69
-0.22
98.52
0.62
4.2
12
19.18
20.05
0.87
104.54
0.88
4.4
0.46
2.7
12
39.86
39.31
-0.55
98.62
1.67
4.3
12
49.77
48.93
-0.84
98.31
1.43
2.9
0.78
1.8
Real
11
0.50
0.94
0.44
188.00
0.80
84.7
Wastewater
10
0.69
0.69
0.00
100.00
0.09
13.3
0.39
47.6
10
1.99
3.00
1.01
150.75
0.38
12.7
10
2.97
3.01
0.04
101.35
0.20
6.6
0.23
7.8
11
14.91
14.52
-0.39
97.38
0.66
4.6
11
19.18
19.26
0.08
100.42
0.77
4.0
0.77
4.6
11
39.86
39.13
-0.73
98.17
1.78
4.6
11
49.77
49.17
-0.60
98.79
2.26
4.6
0.93
2.1
23
Table
6
Precision, Bias, and Matrix Recovery for
Fluoride
Matrix
# of
True
Mean
Bias
vs
Recovery
Interlab
Intortab
Single
Analyst
Values
Value
Result
True
vs
True
Std Dev
%RSD
Operator
%RSD
Value
Value
S(t)
Std 0ev,
S(o)
Reagent
10
0.50
0.51
0.01
102.00
11.00
11.4
Water
10
0.71
0.73
0.02
102.82
7.90
8.1
0.02
2.9
10
2.00
2.05
0.05
102.50
3.60
3.7
10
3.00
2.96
-0.04
98.67
4.40
4.6
0.09
3.4
10
6.99
7.02
0.03
100.43
5.40
5.6
10
9.99
9.79
-0.20
98.00
4.60
4.8
0.13
1.6
10
19.98
19.60
-0.38
98.10
3.80
3.9
10
24.99
24.51
-0.48
98.08
4.80
4.9
0.74
3.4
Substitute
10
0.50
0.50
0.00
100.00
0.09
18.0
Wastewater
10
0.71
0.71
0.00
100.00
0.09
12.0
0.01
2.3
10
2.00
1.98
-0.02
99.00
0.12
6.0
10
3.00
2.94
-0.06
98.00
0.10
3.4
0.06
2.6
10
6.99
6.92
-0.07
99.00
0.28
4.1
9
9.99
9.94
-0.05
99.50
0.46
4.7
0.28
3.3
10
19.98
19.67
-0.31
98.45
0.94
4.8
10
24.99
24.78
-0.21
99.16
1.09
4.4
0.63
2.8
Drinking
13
0.50
0.48
-0.02
96.00
0.06
12.9
Water
13
0.71
0.68
-0.03
95.77
0.06
9.5
0.02
3.4
13
2.00
1.96
-0.04
98.00
0.08
3.9
13
3.00
2.90
-0.10
96.67
0.10
3.4
0.08
3.5
13
6.99
6.91
-0.08
98.86
0.25
3.6
13
9.99
9.91
-0.08
99.20
0.37
3.7
0.18
2.2
13
19.98
19.94
-0.04
99.80
0.68
3.4
12
24.99
24.27
-0.72
97.12
1.63
6.7
1.30
5.9
“Real
11
0.50
0.47
-0.03
94.00
0.08
16.9
Wastewater
11
0.71
0.68
-0.03
95.77
0.08
11.7
0.04
7.6
11
2.00
1.96
-0.04
98.00
0.12
6.3
11
3.00
2.93
-0.07
97.67
0.18
6.2
0.09
3.5
11
6.99
6.85
-0.14
98.00
0.26
3.8
10
9.99
9.56
-0.43
95.70
0.73
7.7
0.44
5.3
11
19.98
20.06
0.08
100.40
1.23
6.1
11
24.99
25.12
0.13
100.52
1.34
5.3
0.32
1.4
24
Table 7
Precision,
Bias, and Matrix
Recovery
for o-Phosphate
Matrix
# of
True
Mean
Bias vs
Recovery
Interlab
Interlab
Single
Analyst
Values
Value
Result
True
vs True
Std
Dev
%RSD
Operator
%RSD
Value
Value
S(t)
Std
Dev, S(o)
Reagent
10
0.50
0.41
-0.09
82.00
0.12
29.5
Water
9
0.69
0.51
-0.18
73.91
0.13
26.6
0.03
7.2
10
1.99
1.88
-0.11
94.47
0.16
8.3
10
2.98
2.76
-0.22
92.62
0.14
4.9
0.08
3.2
10
14.86
14.93
0.07
100.47
0.64
4.3
9
19.80
19.76
-0.04
99.80
1.00
5.1
0.85
4.9
10
39.60
39.79
0.19
100.48
1.38
3.5
10
49.51
50.10
0.59
101.19
1.76
3.5
0.72
1.6
Substitute
11
0.50
0.49
-0.01
98.00
0.15
30.0
Wastewater
10
0.69
0.59
-0.10
85.51
0.17
28.8
0.13
24.4
11
1.99
1.92
-0.07
96.48
0.28
14.6
10
2.98
2.89
-0.09
96.98
0.22
7.6
0.18
7.5
11
14.86
15.31
0.45
103.03
1.74
11.4
11
19.80
19.78
-0.02
99.90
1.16
5.9
0.84
4.8
11
39.60
39.58
-0.02
99.95
2.72
6.9
11
49.51
49.19
-0.32
99.35
3.98
8.1
2.18
4.9
Drinking
12
0.50
0.46
-0.04
92.00
0.14
30.0
Water
13
0.69
0.55
-0.14
79.71
0.20
36.3
0.07
13.4
13
1.99
1.89
-0.10
94.97
0.22
11.9
13
2.98
2.87
-0.11
96.31
0.24
8.5
0.07
2.8
12
14.86
15.09
0.23
101.55
0.91
6.1
13
19.80
20.28
0.48
102.42
0.96
4.7
1.06
6.0
13
39.60
40.37
0.77
101.94
2.15
5.3
13
49.51
50.75
1.24
102.50
3.14
6.2
1.03
2.3
ReaI
11
0.50
0.43
-0.07
86.00
0.17
39.1
Wastewater
11
0.69
0.53
-0.16
76.81
0.24
46.5
0.12
25.8
11
1.99
1.72
-0.27
85.43
0.27
15.8
11
2.98
2.52
-0.46
84.56
0.48
19.2
0.30
14.0
11
14.86
14.93
0.07
100.47
0.91
6.1
11
19.80
19.90
0.10
100.51
1.35
6.8
0.91
5.2
11
39.60
38.98
-0.62
98.43
1.45
3.7
10
49.51
48.26
-1.25
97.48
1.80
3.7
0.82
1.9
All
data
determined as spike recovery from ASTM method
validation and EPA
Tier 3 Criteria
Reagent
water
(RW) data between
0.5
and
50
mgIL, except
Fluoride
0.5
and
25 mg/L
consisting
of 4 Youden Pairs
Drinking
(DW) and
Wastewater (WW) data
between 2 and
50 mg/L except Fluoride
2 and 25 mglL
consisting of
3
Youden
Pairs
RSD
= %Relative
Standard
Deviation;
(std dev
I
mean)(1 00)
LL = Lower
Limit
of
%Recovery
UL
= Upper Limit of
%Recovery
RPD
= Relative
%
Difference between
MSD
25
Table
8
QC
Acceptance Criteria
Analyte
Matrix
Precision
Average
Initial
Ongoing
MSIMSD
MS/MSD
%RSD
%Recovery
LL-UL
LL-UL
LL-UL
RPD
Chloride
RW
6.30
98.5
90.8- 106.2
88.7- 108.3
89.4 - 107.5
12.0
DW
10.00
97.0
84.0- 110.0
81.1 - 1130
81.9-
112.5
18.6
WW
7.00
92.8
83.0- 102.6
81.4-
104.2
81.8 - 103.8
13.2
Bromide
RW
10.10
99.7
92.2
- 107.2
86.7 - 112.7
88.5
- 111.0
19.2
DW
12.70
95.8
85.9- 105.6
79.8-
111.8
81.8- 109.8
23.3
WW
14.40
99.2
87.2
- 111.2
80.1
- 118.3
82.4 - 116.0
26.9
Nitrite
RW
6.40
100.6
95.1 - 106.0
91.9- 109.2
92.8- 108.3
12.1
DW
4.30
99.6
92.4 - 106.7
91.5
- 107.7
91.8
- 107.4
8.1
WW
4.90
98.9
91.3-
106.5
90.2-
107.6
90.5-
107.3
9.2
Sulfate
RW
9.40
100.4
90.9- 109.9
86.9- 113.9
88.2- 112.6
17.9
DW
16.1
95.6
82.6- 108.6
74.9-
116.2
77.5- 113.7
29.7
WW
19.60
95.3
78.9 - 111.7
70.1
- 120.5
72.6- 118.0
36.9
Nitrate
RW
8.40
99.5
93.1 - 105.9
88.6-
110.4
90.0-
108.9
15.9
DW
9.40
100.2
93.0- 107.4
88.0- 112.4
89.4- 111.0
17.4
wW
6.70
99.1
90.7
-
107.6
88.6
- 109.7
89.2
- 109.1
12.4
Fluoride
RW
7.90
99.5
92.2-
106.7
88.7-
110.3
89.8-
109.1
14.9
DW
4.86
98.3
91.9
- 104.8
90.5 - 106.2
90.9
- 105.7
9.0
WW
7.90
98.5
90.0-
107.1
87.0-
110.1
88.0- 109.1
14.7
Phosphate
RW
10.60
98.2
91.9
- 104.5
85.4 - 111.0
87.4
- 109.0
20.1
DW
9.40
100.2
89.3
- 111.1
85.8 - 114.6
87.0 - 113.4
17.4
WW
16.90
94.6
81.5-107.7
73.5-115.8
76.1 -113.1
31.5
26
Appendix
B
(Non-mandatory
Information)
B.l Suggested Background
References
Bl .1 EPA
Method
6500,
“Dissolved Inorganic
Anions in Aqueous Matrices
by
Capillary
Ion
Electrophoresis”, SW846, Rev
0, January
1998.
Bi
.2
Method 4140, “Inorganic
Anions by Capillary
Ion Electrophoresis”, Standard
Methods for
the Examination of
Water
and
Wastewater,
20
Edition, 1998,
p
4-12
to
4-20.
Bi
.3
Krol,
Benvenuti,
and
Romano, “Ion Analysis
Methods
for IC and
CIA and
Practical
Aspects of Capillary Ion Analysis Theory”,
Waters Corp. Lit Code WT-139,
1998.
Bi .4
Jandik, P., Bonn,
G.,
“Capillary Electrophoresis
of Small Molecules and Ions”,
VCH
Publishers,
1993
Bi
.5
Romano,
J.,
Krol,
J, “Capillary Ion
Electrophoresis,
An Environmental Method
for the
Determination
of Anions in Water”,
J. of
Chromatography,
Vol.
640, 1993,
p.
403.
B1
.6
Romano, J., “Capillary
Ion
Analysis:
A Method
for
Determining
Ions in Water
and
Solid
Waste
Leachates”, Amer. Lab.,
May 1993,
p.
48.
B1 .7 Jones, W., “Method Development
Approaches for Ion
Electrophoresis”,
J.of
Chromatography,
Vol. 640, 1993,
p.
387.
Bi
.8
Jones, W.,
Jandik,
P., “Various Approaches
to Analysis
of Difficult
Sample
Matrices for Anions using Capillary
Electrophoresis”,
J. of
Chromatography,
Vol.
608,
1992,
p. 385.
Bi .9
Bondoux,
G., Jandik, P., Jones,
W., “New Approaches
to the Analysis
of Low
Level of
Anions
in Water”, J. of Chromatography,
Vol.
602, 1992,
p.
79.
B1.10
Jandik, P., Jones,
W., Weston,
A.,
Brown,
P.,”Electrophoretic
Capillary
Ion
Analysis: Origins, Principles,
and Applications”,
LCGC, Vol.
9, Number
9, 1991,
p.
634.
Bi .11 Romano,
J.,
Jackson,
P., “Optimization
of Inorganic
Capillary
Electrophoresis
for the Analysis of Anionic
Solutes in Real
Samples”,
J. of Chromatography,
Vol.
546,1991,
p.
411.
Bi .12
Jandik,
P., Jones,
W., “Optimization
of
Detection
Sensitivity
in the
Capillary
Electrophoresis of Inorganic
Anions”, J
of Chromatography,
Vol.
546, 1991,
p.
431.
B1 .13 Jandik, P., Jones,
W., “Controlled
Changes
of Selectivity
in the
Separation
of
Ions
by
Capillary Electrophoresis”,
J.
of
Chromatography,
Vol.
546, 1991,
p
445.
Bi .14
Foret,
R.,
et.al.,
“Indirect
Photometric Detection
in
Capillary Zone
Electrophoresis”,
J.
of Chromatography,
Vol. 470,
1989,
p.
299.
B1 .15 Hjerte’n, S. et. al., “Carrier-free
Zone Electrophoresis,
Displacement
Electrophoresis
and
Isoelectric Focusing
in an Electrophoresis
Apparatus”,
J.of
Chromatography,
Vol.
403, 1987,
p.
47.
Bi
.16 Serwe, M., “New ASTM
Standard:
Recommended
Operating
Conditions
for the
Agilent CE”, Agilent
Technologies
Application Brief,
Publication
Number
5968-
8660E.
27
Appendix
C
Capillary
Ion
Electrophoresis
Initial
Demonstration
of
Performance
Single
Operator
General
Inorganic
Anion
&
Organic
Acid
Analysis
with
Indirect
UV
Detection
Basis
for EPA
Method
6500,
ASTM
D6508,
and
Standard
Methods
4140
The
performance
data
given
in
this
appendix
was
provided
in
the
collaborative
instruction
booklet
to
evaluate
initial
demonstration
of performance
required
by
the
collaborative
design.
D
E
C,,
Analysis
Conditions:
Electrolyte:
Capillary:
Temperature:
Power
Supply:
Voltage:
Current:
Sampling:
Detection:
Time
Constant:
Sampling
Rate:
Analyte
MT:
Quantitation:
lonSelect
High
Mobility
Anion
Electrolyte,
P/N
49385
75 .tm
(Id)
x 375
jm (od)
x 60
cm
(length)
25°C
(5°C
Above
Ambient)
Negative
15 kV
14
± 1 jiA
(Use
Constant
Current
for Analysis)
Hydrostatic
for
30
Seconds
Indirect
UV
at
254 nm,
Hg
Lamp,
185
or
254
nm Window
0.3
Seconds,
or less
20. Data
Points
per Second
Mid-Point
of
Analyte
Peak
Width
at
Baseline
Time
Corrected
Peak
Area
(Peak
Area
I
MT)
6
3
4
PPM
Standards
1
Chloride
= 2
2
Bromide
=4
3
Nitrite
=
4
4 Sulfate
= 4
5
Nitrate
=
4
9
6
Oxalate
= 5
7
Fluoride
=
1
8 Formate
= 5
9
Phosphate
=
4
10 Bicarbonate
11 Acetate
= 5
10
3.000
3.500
Minutes
4.000
28
Millennium
Data
Processing
Method:
CIE
Processing
Method
using
Mid-Point
of Peak
Width
for
Migration
Time
Integration
Peak Width
=
2.25
- 3.00
Threshold
= 100 ±25
Mm
Area
= 100
Mm
Height
= 50
Inhibit
lntg. =Oto3min
Calibration
Averaging
=
None
MT
Window
= 2%
Update
MT
=
Average Standards
Peak
Match
= First
for
Chloride
(Cl is always
first in the
pherogram,
use as a
ref
peak)
Cl MT Window
=10%
Other
Analytes
= Closest
Quantitate
By = Time
Corrected
Peak
Area
Fit Type
= Linear
Through
Zero
Report
Analyte
Name
Analyte
Migration
Time
Analyte
Migration
Time
Ratio
(respect
to Cl Ref
Peak)
Peak
Area
Time
Corrected
Peak Area
Amounts
Use
fresh
electrolyte
daily;
recalibrate
with
every change
in
electrolyte.
Clear
previous
calibration
(in
Quick Set
Page) before
recalibration.
Do Not use
analyte
peak
height
for
quantitation
due
to
asymmetrical
peak shapes.
Method
Validation:
The single
operator
performance
given
below
using
the
ASTM
validation
design is
intended
as
a basis
to
evaluate
Initial
Demonstration
of
Performance.
Individual
Youden
Pair Standard,
in ppm
1
234
56
7
8
Cl
0.7
2.0
3.0
15.0
40.0
20.0
50.0
0.5
Br
2.0
3.0
15.0
40.0
20.0
50.0
0.7
0.5
N02
3.0
40.0
20.0
15.0
50.0
0.5
2.0
0.7
S04
40.0
50.0
0.5
0.7
2.0
3.0
15.0
20.0
NOs
15.0
20.0
40.0
50.0
0.5
0.7
2.0
3.0
F
2.0
0.7
0.5
3.0
10.0
7.0
20.0
25.0
P04
50.0
40.0
20.0
0.5
3.0
2.0
0.7
15.0
C
C
C
G)
ct
C
29
Cl
Cl
R
2
=
0.9996
SO
4
R
2
=
0.9998
Br
R
2
=
0.9995
Method
Linearity:
10
7.5
Cs
52
CO
w
m
.
CO
—
0)
CJ)
0
2.5
E
I-
0
10
7 .5
Cs
Cs
2.5
0
10
20
30
40
50
ppm Anion
F
A
2
0.9985
P0
4
A2
= 0.9996
P04
10
3
Data Points
per
Concentration
U sing
Va lid ation
S ta nda rd
a
0
10
20
ppm
Anion
30
40
50
7
.5
NO
2
R
2
=
0.9996
N0 R
2
=
0.9992
5
CO
Cs
Cs
Cs
0
0
Cs
0
Cs
0
C.)
Cs
E
I-
en
0
C
Cs
4)
0
I.
2.5
N02
0
NO
0
10
20
ppm
Anion
30
40
50
30
Method
Detection
Limits:
D
2
[K)
d
Minutes
Seven
replicates
of the
above 100 ppb
anion
standard
were used
to calculate time
corrected
peak
area
precision.
Using
EPA
and Standard Methods
protocols,
the detection
limits,
as
ppb,
for these
analytes are:
Chloride
=46
Nitrate
=84
Bromide =90
Fluoride =20
Nitrite =72
Phosphate
=41
Sulfate
=32
This
method has been
validated between
0.1
to
50
ppm.
Quantitation
below
0.1
ppm
is not
advised.
100 ppb Anion Standard
CT
S04
F
N02
NOa
P04
3.60 0
31
Migration
Time
Reproducibility:
Use
mid-point
of
analyte
peak
width
at
the
baseline
as
the
analyte migration
time
determinant.
Data
given
as average
absolute
migration
time
for
each
validation
standard analyzed
in
triplicate.
Analyte
Cl
Br
N02
S04
N03
F
P04
1
3.132
3.226
3.275
3.405
3.502
3.761
3.906
2
3.147
3.239
3.298
3.431
3.517
3.779
3.931
3
3.138
3.231
3.283
3.411
3.497
3.771
3.925
4
3.158
3.244
3.307
3.434
3.510
3.781
3.963
5
3.184
3.271
3.331
3.435
3.551
3.787
3.981
6
3.171
3.260
3.312
3.418
3.537
3.776
3.964
7.
3.191
3.272
3.315
3.437
3.544
3.773
3.978
>
8
3.152
3.248
3.294
3.418
3.526
3.739
3.954
Std
Dev
0.021
0.015
0.018
0.012
0.20
0.015
0.027
%RSD
0.67%
0.46%
0.55%
0.36%
0.56%
0.40%
0.68%
Average
Standard
Deviation
= 0.018
mm = 1.1
sec
Average
%RSD
of Analyte
Migration
Time
= 0.53%
Quantitation
Precision:
Time
Corrected
Peak Area
Precision,
given
as %RSD,
based upon
3
samplings
per
concentration.
Analyte
Cl
Br
N02
S04
N03
F
P04
0.1
12.36
18.89
16.19
13.25
23.13
9.82
14.00
0.5
10.51
20.00
3.90
2.25
2.18
2.03
7.71
0.7
1.23
13.36
2.01
2.95
0.37
2.72
4.41
2
0.32
3.76
4.14
1.79
2.17
0.73
1.91
C.,
g
3
0.63
1.80
1.72
1.70
0.58
0.98
2.70
C.)
E
15
0.43
0.27
0.48
0.07
0.36
0.15
1.37
° 20
0.45
0.66
0.17
0.13
0.88
0.16
0.81
40
0.36
0.56
0.36
0.46
0.58
0.47
50
0.45
0.51
0.48
0.16
0.46
0.46
32
Quantitation Accuracy:
Used
a Certified
Performance Evaluation
Standard diluted 1:100 with Dl water.
Amounts
based upon multi-point calibration curve prepared from
certified standards.
Analyte
Cl
N02
S04
N03
F
P04
Performance
True
Evaluation
Value
43.00
1.77
37.20
15.37
2.69
6.29
Standard
in ppm
Official
Measured
43.30
1.77
37.00
15.42
2.75
6.38
Anion
Mean
Methods
Measured
3.09
0.07
2.24
1.15
0.26
0.21
WetChem&IC
StdDev
CIA
Using
Ave
CIA
43.34
1.64
37.11
14.41
2.64
6.34
Chromate
n=1
8
Electrolyte
CIA/Mean
1.003
0.927
1.003
0.935
0.959
0.993
CIA/TrueValue
1.008
0.927
0.996
0.938
0.981
1.008
A
CIA/True
Value,
or Mean
= 1.000
indicates
perfect agreement
between CIA and
official
anion methods.
Method
Recovery:
A Certified Performance
Evaluation Standard
(PES) was diluted
1:100
with
Typical
Drinking
Water (DW). Amounts
based upon multi-point calibration
curve
prepared from
certified
standards.
Analyte
CI
N02
S04
N03
F
P04
Drinking Water
24.72
±
Not
7.99
±
0.36
±
Not
Not
n=3,
as ppm
0.18
Detected
0.07
0.05
Detected
Detected
Amount %RSD
0.73%
0.91%
13.3%
Performance
43.00
1.77
37.20
15.37
2.69
6.29
Evaluation Std
DW + PES
66.57±
1.74±
45.19±
15.42±
2.62±
n=3;
as
ppm
0.34
0.03
0.17
0.12
0.07
0.31
Amount %RSD
0.51%
1.85%
0.38%
0.79%
2.69%
5.52%
%
Recovery
97.9%
98.3%
100.2%
98.1%
97.4%
88.2%
33
Fig. 3
Electropherogram
of
Substitute
Wastewater
Fig.
5 Electropherogram of Municipal
Wastewater
Treatment
Plant
Discharge
Fig. 4
Electropherogram
of Drinking
Water
Fig.
6 Electropherogram
of
Industrial
Wastewater
Fig.
1 Electropherogram
of
Mixed
Anion
Working
Solution
and Added
Common
Organic Acids
Fig.
2
Electropherogram
of 0.2
mg/L
Anions
Used to
Determine
MDL
1
Chbide=2
7 F1uoiic
= I
3
4
2 BrolTicie
= 4
8 Formate
= 5
3 Nitrite
= 4
9
Phoophate
= 4
8
4Sulfate =4
loCaboiiate
5 Ndre
= 4
11
Acetate
=5
±AJLRLI
I Chloride
2
Sronide
3
Nftrite
4
Sulfate
S Nitre
6
Fluoride
7 Pho,hate
5
C,)
C
4
3.000
3.500
4000
4.500
Mi,utes
3.200
3.400
Mrrutes
3.600
C
Mbns
ii
mo/l_
120
0utlon
1 Chloride
= 24.2
2Sulfate
=3.77
3
Phosphate
= 0.89
4 Carbonate
= Natral
U)
a
Mensnmorl_
fk,0uiton
1
Chloride = 20.2
2
Sulfate
=
7.5
3Nitrae
=1.6
4 Fluoride
= 0.C6
5
Carbonate
= NatLrat
3
S
4.500
Anions
ii
mO/L,
rb
Diution
1
Chloride
= 93.3
2
Nitrite
=
0.46
3
Sulfate
= 60.3
4
Nitre
=
40.8
5
Carbonate
= Natrsal
3
D
00
a
Anions
ri
ma&. te)
Diution
I Chloride
= 2.0
2 Nitrite
= 1.6
3
Sulfate
=34.7
4 Nltre
=
las
5
Formale
<
0.05
6 Ftrosphate
= 12.3
7
Carbonate
= Natral
4
3.500
MinUtes
3.000
4.000
4.500
Mioutes
4.000
4.500
34
Fig.
7 Pictorial
Diagram
of Anion
Mobility and
ElectroOsomotic
Flow
Modifier
CMhod.
HIebMthllItyAnIon
(
“.__j
(
Low MobSfty MIon
),
I
J
N.otrol. &W.,
\
)
—CAc.D
soe—).
1r4.o5on
Sid.
Daon
sia.
e
a
a
a
a
AAA
Ae
e
e
a eee
a
a net
dase
n badground
soibance.
T1e
charge in
soibance
is
drectly
retated
to Nietyte concaitration.
Fig. 8
Selectivity
Diagram of
Anion
Mobility
Using
Capillary
Ion Electrophoresis
——sç—y----—-—--—I
—f
—
—
O
0.
0-
0
0.
o
0.
OH
i:i:.
N&-..-’---v-
•‘ii+
N.N
+
Fig. 9
Pictorial Diagram
of Indirect
UV Detection
Migration
Time
MT=
0
Hkih Mobility
Low Mohlhtv
MT >7
mm
Anions
Anions
Fig.
10
General
Hardware
Schematic
of a
Capillary
Ion Electrophoresis
System
see
a ee
AA
As
e e
a
eee
ee
a a
ee
eAAA
As a
a
a e a
e
a
e
e e
a e
eAAA
As e
e e e e
e
e
a,
a,
E